

")








Abernethy malformation: A case report
- Authors: Panyukova A.V.1, Sinitsyn V.E.1, Mershina E.A.1, Rucheva N.A.2
-
Affiliations:
- Lomonosov Moscow State University, Medical Research and Educational Center
- V.I. Shumakov National Medical Research Center of Transplantology and Artificial Organs
- Issue: Vol 4, No 2 (2023)
- Pages: 226-237
- Section: Case reports
- Submitted: 06.03.2023
- Accepted: 03.04.2023
- Published: 12.07.2023
- URL: https://jdigitaldiagnostics.com/DD/article/view/289714
- DOI: https://doi.org/10.17816/DD289714
- ID: 289714

Cite item

Abstract
Congenital portosystemic shunts are rare congenital vascular malformations characterized by a partial or complete portal blood diversion into the systemic circulation. Congenital extrahepatic portosystemic shunts, known as Abernethy malformations, pose a diagnostic challenge due to their low incidence and clinical presentations.
A 15-year-old male with a history of chronic epigastric pain and nausea, high arterial blood pressure, recurrent nose bleeds, chest pain, dizziness, dyspnea, low exercise tolerance, hematochezia, and itching was diagnosed with Abernethy malformation type Ib. Imaging studies revealed a dilated portal vein conduit flowing into the inferior vena cava, bypassing the porta hepatis. Multiple liver nodules, heart chamber dilatation, myocardial hypertrophy, and pulmonary hypertension were also discovered. Following multidisciplinary panel meetings, liver transplantation was advised due to the severity of the patient’s symptoms and shunt anatomy.
Furthermore, diagnostic algorithms and other treatment options are discussed.
Full Text
INTRODUCTION
Congenital portosystemic shunts (CPSS) are rare congenital anomalies associated with partial or complete portal blood diversion into the systemic circulation. The estimated CPSS incidence is 1:30,000 births and 1:50,000 for those that persist beyond early life [7]. The classification of CPSS is complex because of the significant variability of vascular anatomy. All CPSSs are divided into intra- and extrahepatic shunts with partial or complete portal blood deprivation [27]. Congenital extrahepatic portosystemic shunts (CEPSS) are termed Abernethy malformation, first documented in 1793 by John Abernethy [1]. However, reported CEPSS cases are limited.
DESCRIPTION OF THE CASE
A 15-year-old male was admitted to the hospital for evaluation of chronic epigastric pain and nausea. He also had episodes of high arterial blood pressure (reaching 160/90 mmHg), recurrent nose bleeds, episodes of chest pain, dizziness, shortness of breath, low exercise tolerance, hematochezia, and long history of itching. His medical history was limited: 12 years prior to admission, portal hypertension was diagnosed (no medical records provided).
Liver function tests showed a mild increase in alanine aminotransferase (59.8 U/L (normal range, 13–50 IU/L)) and increased aspartate aminotransferase (67.1 U/L (15–46 IU/L)), gamma-glutamyl transferase (91 U/L (2–42 U/L)), alkaline phosphatase (316 U/L (52–171 U/L)), total bilirubin (39.2 μmol/L (3.4–17.1 μmol/L)), and direct bilirubin (12.5 μmol/L (0–5 μmol/L)); albumin was slightly decreased (40.2 g/L (41–55 g/L)). Routine blood tests and coagulation studies were normal. Further, his serum BUN, and creatinine were within reference ranges.
Transthoracic echocardiogram revealed heart chamber dilatation, myocardial hypertrophy (left ventricular wall thickness, 1.6 cm), and systolic pulmonary hypertension (pulmonary artery systolic pressure [PASP], 40 mmHg). Aortic ectasia (diameter at the level of the fibrous ring, 3.4 cm; sinuses of Valsalva, 5.1 cm; and ascending aorta, 4.0 cm) was observed. No left ventricular outflow tract stenosis or ventricular wall hypokinesia were found; left ventricular function was preserved.
Abdominal ultrasound (US) showed an enlarged liver with multiple nodules, changes in parenchymal structure, and signs of fibrosis. No prominent portal venous trunk or branches at the porta hepatis were noted. The hepatic vascular pattern was deformed with stenosis of the hepatic veins. Further findings were portal hypertension and moderate spleen enlargement.
To evaluate liver nodules, alpha-fetoprotein (AFP) tumor marker test was ordered. The AFP concentration was normal (1.72 IU/ml). Additional imaging studies were performed to confirm the diagnosis and to clarify the vascular anatomy.
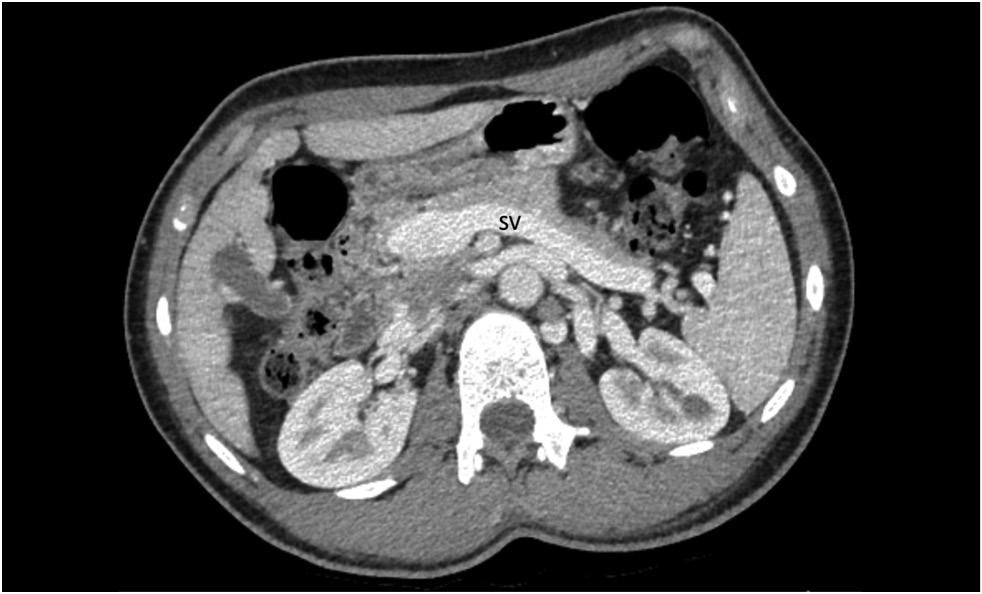
Contrast-enhanced abdominal computed tomography (CT) with multiplanar reconstruction revealed that the splenic (12 mm in diameter (Figure 1)) and superior mesenteric veins fused together, forming a portal vein conduit dilated to 28 mm in diameter (Figures 2 and 3), flowing directly into the inferior vena cava (IVC), bypassing the porta hepatis (Figure 4). Moreover, moderate liver and spleen enlargement and weak heterogeneous contrast enhancement of the liver parenchyma were noted. The findings were consistent with Abernethy malformation type Ib.

Fig. 1. Contrast-enhanced CT, portal phase, axial view. Dilated splenic vein (SV).

Fig. 2. Contrast-enhanced CT, portal phase, coronal view. Splenic (SV) and superior mesenteric (SMV) veins fused together, forming a portal vein conduit (white arrow).

Fig. 3. Contrast-enhanced CT, portal phase, axial view. Portal vein conduit (white arrow).

Fig. 4. Contrast-enhanced CT, portal phase, coronal view. Portal vein conduit flowing directly into the IVC (white arrow), enlarged liver with heterogeneous parenchymal enhancement.
CT pulmonary angiogram showed no abnormal vascular shunts, but confirmed pulmonary trunk dilatation (diameter, 40 mm) (Figure 5), heart chamber dilatation, and myocardial hypertrophy (Figure 6).

Fig. 5. CT pulmonary angiography, axial view. Pulmonary trunk dilatation.

Fig. 6. CT pulmonary angiography, axial view. Myocardial hypertrophy.
Owing to the low effectiveness of conservative treatment, severity of symptoms, and shunt anatomy, liver transplantation was recommended after multidisciplinary panel consultations. Currently, the patient is waiting for surgical intervention.
DISCUSSION
Mechanisms
The etiology and development of congenital and acquired portosystemic shunts differ significantly. CEPSSs occur because of abnormal formation or involution of the fetal vasculature; acquired shunts are secondary to liver diseases [27]. In the literature, there are two dominant theories of CEPSS formation: congenital malformations and anomalies of the ductus venosus.
The development of the portal system is complex and occurs between weeks 4 and 10 of embryonic life. The systemic venous system results from the embryonic anterior and posterior cardinal veins. The portal venous system forms from the vitelline veins, which carry blood from the yolk sac to the sinus venosus [13]. If portal system development is disrupted, CEPSS occurs. This variant is closely associated with combined congenital pathologies. According to the study of Bernard et al., congenital heart disease was the most frequently observed concomitant pathology (in 45/265 cases); other recorded malformations included abnormalities of the kidneys, bile ducts (including biliary atresia), digestive system, bones, and brain [7].
Another discussed mechanism is the absence of the functioning fetal ductus venosus due to anatomical defects or occlusion. In a normal fetus, the ductus venosus shunts blood from the umbilical vein to the IVC, bypassing the liver. Naturally, functional closure occurs within the first minutes of birth and structural closure during the first weeks of life in most full-term neonates [8]. The umbilical vein and ductus venosus anatomically close during the first months of life and become the ligamentum teres and ligamentum venosum, respectively [13]. Pathologies of the ductus venosus in the fetus can stimulate the formation of abnormal vessels. These abnormal vessels may persist and develop into abnormal shunts, resulting in hypoplasia of the portal venous system. The absence of the ductus venosus has been reported in some cases of CEPSS [5, 12].
Classification
A widely used classification of CEPSS is the classification system introduced by Morgan and Superina in 1994 (Table 1) [23]. According to this classification, Abernethy malformation is divided into two types depending on the patency of the intrahepatic portal system. Type 1 is defined as a complete portosystemic shunt, whereas type 2 is described as partial blood shunting to systemic veins with a certain degree of portal system development (Figure 7). Different treatment options are available depending on the CEPSS type [35].
Table 1. Classification System for portasystemic anomalies by Morgen and Superina [23].
Type I | Liver not perfused with portal blood — total shunt |
Ia: SMV and splenic vein do not joint to form confluence | |
Ib: SMV and splenic vein join to form confluence | |
Type II | The liver perfused with portal blood — partial shunt (e.g., portal-hepatic venous anastomoses) |
IIa: congenital | |
IIb: acquired |
Note. SMV — superior mesenteric vein.

Fig. 7. Normal portal vein anatomy and shunt classification. IVC — inferior vena cava; PV – portal vein; SV — splenic vein; SMV — superior mesenteric vein; a — normal PV anatomy; b — CEPSS type Ia; c — CEPSS type Ib; and d — CEPSS type II.
Clinical manifestations and complications
Clinical presentations are variable and depend on the ratio of blood flow through the shunt. Manifestations vary from accidental findings in asymptomatic adult patients [26, 32] to complex congenital malformations [36], severe hypoxemia [31], encephalopathy [21, 22], or liver tumors [33]. Most patients present with nonspecific symptoms, such as acute hepatic decompensation or cirrhosis. According to Lin et al., data of 703 patients with CEPSS extracted from 451 articles revealed that majority of the reported patients with Abernethy malformation were children or young adults <18 years old [20]. Severe congenital pathologies with a higher degree of blood shunting are usually diagnosed at a younger age, whereas patients with partial blood shunting can remain asymptomatic till adulthood.
In the early neonatal period, galactosemia, diagnosed during routine screening, can be the first sign of CEPSS. Galactose is metabolized in the liver by the GALT enzyme to glucose. However, in children with CEPSS, galactose bypasses the liver, resulting in increased levels in the systemic circulation [14, 29]. According to several researchers, hypergalactosemia is present in up to 70% of newborns with CPSS [7]. Other potential symptoms in the early neonatal period are growth restriction, neonatal cholestasis, and hepatic encephalopathy [28].
In patients with milder pathology, CEPSS can go unnoticed until adulthood. The presentation may be due to symptoms related to hepatic encephalopathy, liver masses, or pulmonary hypertension.
Subclinical hepatic encephalopathy is observed in up to 30% of patients with CEPSS [11]. Portal blood shunting causes increased ammonium levels in the systemic blood flow. Blood ammonia produced in the gastrointestinal tract bypasses the liver and flows directly into the IVC. Astrocytes metabolize ammonium to glutamine, which has toxic effects on the brain [21]. Hyperammonemia may present without encephalopathy, particularly at younger ages. Clinical encephalopathy is more common in older patients, probably due to lower compensatory abilities [22]. Diagnosis in such cases can be difficult because of the low specificity of symptoms [2, 3, 21]. Elevated serum ammonia concentration without evidence of liver cirrhosis should prompt further investigations for extrahepatic shunts.
Patients with CPSS are prone to developing multiple liver tumors. The literature on histological changes in the liver parenchyma in patients with CPSS is limited. De Vito et al. described a case series of 22 patients with CPSS, including 19 patients with CEPSS, who were diagnosed and managed in their institution for 15 years [10]. According to their results, the most characteristic histological findings in peripheral liver parenchyma included the presence of portal prominent thin-walled channels, arterial-biliary dyads, increased arterial profiles in the portal tracts and lobule, and frequent lack of the physiological periportal-vacuolated hepatocytes in children.
The pathophysiology of hepatic tumor in patients with CEPSS remains unclear. One of the mechanisms is attributed to reduced liver regeneration abilities. Low portal blood flow leads to a decrease in the delivery of insulin and glucagon to hepatocytes, making them more vulnerable to damage and neoplasm development [17]. Moreover, increased hepatic arterial blood flow can be associated with parenchymal cell de-differentiation [33].
Nodular liver lesions are common findings in different types of Abernethy malformation. In most cases, liver nodules are benign, and include focal nodular hyperplasia, hepatic adenomas, and regenerative nodules. Most patients are asymptomatic, although some patients present with an abdominal mass. In our case, liver nodules were accidentally found during abdominal US.
However, not all liver masses are benign. Type I Abernethy malformation is associated with hepatoblastoma and hepatocellular carcinoma (HCC) [9, 16]. Hepatoblastoma is a rare tumor seen in children with CEPSS. These tumors have an unfavorable prognosis. Majority of the described cases were lethal [9, 17]. Hepatocellular carcinomas more often develop in adults, although Benedict et al. published a case of a 12-month-old male with histologically and immunohistochemically confirmed HCC [6]. Diagnosis can be complicated as some of the reported lesions have controversial radiological features and can be mistaken for benign masses [32]; biopsy is usually required. Liver transplant is a treatment option [25].
In some CEPSS cases, patients present with signs of pulmonary hypertension: shortness of breath and dyspnea [19, 24]. Severe pulmonary hypertension can lead to cardiogenic syncope due to decreased preload and low cerebral perfusion [20]. In our case, pulmonary hypertension was diagnosed.
Hepatopulmonary syndrome occurs in patients with liver disease accompanied by portal hypertension. Vasoactive mediators from the intestine bypass the hepatic circulation through the portosystemic shunt, flowing directly to the pulmonary vascular bed, causing imbalance between vasodilation and vasoconstriction substances, inducing pulmonary hypertension [35]. The correction of hepatic vascular anomalies is curative.
Diagnosis and treatment
There are currently no published guidelines for the diagnosis and treatment of CEPSS. Based on the results of the multicenter international study that included 66 patients, Baiges et al. have proposed a management algorithm for patients with CEPSS (Figure 8) [4].

Fig. 8. A congenital extrahepatic portosystemic shunt management algorithm proposed by Baiges et al. [4]. CEPS — congenital extrahepatic portosystemic; CT — computed tomography; HCC — hepatocellular carcinoma; HE — hepatic encephalopathy; MRI — magnetic resonance imaging; US — ultrasound.
In our case, Abernethy malformation was suspected in the abdominal US. Generally, US signs of CEPSS include portal trunk absence or hypoplasia, solid focal lesions in the liver parenchyma, deficiencies of the intrahepatic portal vessels and flow signals, and hepatic artery hypertrophy [30]. Anomalies identified by the US should be further confirmed with other imaging modalities, such as CT, or MR angiography. Contrast-enhanced CT provides essential information about shunt size, orientation, and type, which helps in choosing the most suitable treatment approach for each patient. Further, it allows to visualize and evaluate concomitant anomalies, including liver masses. MR angiography is a reliable and noninvasive modality for visualizing hepatic vascular anatomy. It is radiation-free and has better soft tissue contrast than CT. Moreover, diffusion-weighted sequences and hepatocyte-specific contrast agents can provide additional valuable information for the evaluation of nodular liver lesions and decision-making.
The therapeutic approach depends on the shunt type and size, severity of symptoms, coexisting anomalies, and related complications. Asymptomatic patients could be medically followed. Given the complication development risks, Kwapisz et al. recommended routine clinical assessments, regular blood work, including liver enzyme and liver function tests, and annual liver imaging for patients with CEPSS [16].
Experience in the treatment of patients with Abernethy malformation remains limited. Based on the reported cases, current treatment options include interventional or surgical shunt closure and liver transplantation. Type I long-term treatment options are limited to the liver transplant with supportive therapy while waiting for surgery. Patients with Type II CEPSS have more therapeutic options depending on the developed complications and associated anomalies. It is possible to ligate or close the portosystemic shunt using interventional angiography (with coils or plugs) [34]. However, interventional closure may cause recurrent hyperammonemia, as has been reported [18].
It may be beneficial to perform the balloon shunt occlusion test to assess the intrahepatic portal system (IHPS) in patients with both types of CEPSS [18]. This test allows to visualize small portal vein branches which cannot be seen on US. Kanazawa et al. proposed a new IHPS classification (mild, moderate, and severe) based on the results of the shunt occlusion test [15]. The IHPS classification correlates with the portal venous pressure under shunt occlusion, histopathological findings, postoperative portal venous flow, and liver regeneration, and is useful for decision-making whether to perform single-stage or two-stage shunt closure or liver transplantation.
CONCLUSION
Abernethy malformation is a rare pathology associated with severe complications and poor outcomes. Owing to the low incidence, unspecific symptoms, involvement of different organ systems, and variable presentations, diagnosis of CEPSS is a challenge. Imaging plays a critical role in the diagnosis and treatment planning. Early identification and individualized treatment approaches are crucial in preventing complications. Long-term follow-up and monitoring for malignancy are required.
ADDITIONAL INFORMATION
Funding source. This study was not supported by any external sources of funding.
Competing interests. The authors declare that they have no competing interests.
Authors’ contribution. All authors made a substantial contribution to the conception of the work, acquisition, analysis, interpretation of data for the work, drafting and revising the work, final approval of the version to be published and agree to be accountable for all aspects of the work.
Consent for publication. Written consent was obtained from the patient's parents for publication of relevant medical information and all of accompanying images within the manuscript in Digital Diagnostics journal.
About the authors
Alexandra V. Panyukova
Lomonosov Moscow State University, Medical Research and Educational Center
Email: panyukovaalexandra@gmail.com
ORCID iD: 0000-0002-5367-280X
Russian Federation, Moscow
Valentin E. Sinitsyn
Lomonosov Moscow State University, Medical Research and Educational Center
Email: vsini@mail.ru
ORCID iD: 0000-0002-5649-2193
SPIN-code: 8449-6590
MD, Dr. Sci. (Med); Professor
Russian Federation, MoscowElena A. Mershina
Lomonosov Moscow State University, Medical Research and Educational Center
Email: elena_mershina@mail.ru
ORCID iD: 0000-0002-1266-4926
SPIN-code: 6897-9641
MD, Cand. Sci. (Med.), Assistant Professor
Russian Federation, MoscowNatalya A. Rucheva
V.I. Shumakov National Medical Research Center of Transplantology and Artificial Organs
Author for correspondence.
Email: rna1969@yandex.ru
ORCID iD: 0000-0002-8063-4462
MD, Cand. Sci. (Med.)
Russian Federation, MoscowReferences
- Bernard O, Franchi-Abella S, Branchereau S, et al. Congenital portosystemic shunts in children: Recognition, evaluation, and management. Seminars Liver Dis. 2012;32(4):273–287. doi: 10.1055/s-0032-1329896
- Papamichail M, Pizanias M, Heaton N. Congenital portosystemic venous shunt. Eur J Pediatrics. 2018;177(3):285–294. doi: 10.1007/s00431-017-3058-x
- Abernethy J. Account of two instances of uncommon formation in the viscera of the human body: From the philosophical transactions of the royal society of London. Med Facts Observations. 1797;(7):100–108.
- Guérin F, Blanc T, Gauthier F, et al. Congenital portosystemic vascular malformations. Seminars Pediatric Sur. 2012;21(3):233–244. doi: 10.1053/j.sempedsurg.2012.05.006
- Born M. The ductus venosus. RoFo Fortschritte Gebiet Rontgenstrahlen Bildgebenden Verfahren. 2021;193(5):521–526. doi: 10.1055/a-1275-0984
- Baller SE, Reinehr M, Haslinger C, et al. Case report of neonatal ductus venosus atresia. J Neonatal-Perinatal Med. 2021;14(2):307–312. doi: 10.3233/NPM-190398
- Franchi-Abella S, Branchereau S, Lambert V, et al. Complications of congenital portosystemic shunts in children: Therapeutic options and outcomes. J Pediatric Gastroenterol Nutrition. 2010;51(3):322–330. doi: 10.1097/MPG.0b013e3181d9cb92
- Morgan G, Superina R. Congenital absence of the portal vein: Two cases and a proposed classification system for portasystemic vascular anomalies. J Pediatric Sur. 1994;29(9):1239–1241. doi: 10.1016/0022-3468(94)90812-5
- Tang H, Song P, Wang Z, et al. A basic understanding of congenital extrahepatic portosystemic shunt: Incidence, mechanism, complications, diagnosis, and treatment. Intractable Rare Dis Res. 2020;9(2):64–70. doi: 10.5582/irdr.2020.03005
- Păcurar D, Dijmărescu I, Dijmărescu AD, et al. A case report on an incidental discovery of congenital portosystemic shunt. Medicine. 2019;98(31):e16679. doi: 10.1097/MD.0000000000016679
- Shah A, Aziz A, Awwad A, et al. Incidental radiological diagnosis of asymptomatic Abernethy malformations: Two case reports. BJR|Case Reports. 2017;3(1):20150496. doi: 10.1259/bjrcr.20220059
- Yangín-Ergon E, Ermis N, Colak R, et al. Abernethy malformation type 2 and biliary atresia coexistence: A rare cause of infantile liver transplant. Euroasian J Hepatogastroenterol. 2018;8(2):163–166. doi: 10.5005/jp-journals-10018-1283
- Sahu MK, Bisoi AK, Chander NC, et al. Abernethy syndrome, a rare cause of hypoxemia: A case report. Ann Pediatric Cardiol. 2015;8(1):64–66. doi: 10.3389/fcvm.2021.784739
- Lux D, Naito A, Harikrishnan S. Congenital extrahepatic portosystemic shunt with progressive myelopathy and encephalopathy. Practical Neurol. 2019;19(4):368–371. doi: 10.1136/practneurol-2018-002111
- Merola E, Cao M, La Starza S. et al. Portosystemic encephalopathy in an 86-year-old patient: A clinical challenge. Acta Gastro-Enterologica Belgica. 2016;79(1):58–59.
- Sharma R, Suddle A, Quaglia A, et al. Congenital extrahepatic portosystemic shunt complicated by the development of hepatocellular carcinoma. Hepatobiliary Pancreatic Dis Int. 2015;14(5):552–557. doi: 10.1016/S1499-3872(15)60418-0
- Lin X, Rao J, Xiang Y, et al. Case report: A rare syncope case caused by abernethy II and a review of the literature. Frontiers Cardiovascular Med. 2022;8:2050. doi: 10.3389/fcvm.2021.784739
- Hasegawa T, Sato T, Ishii T, et al. Oral sodium phenylbutyrate for hyperammonemia associated with congenital portosystemic shunt: A case report. J Pediatric Endocrinol Metabolism. 2021;34(3):407–410. doi: 10.1515/jpem-2020-0603
- Peček J, Fister P, Homan M. Abernethy syndrome in Slovenian children: Five case reports and review of literature. World J Gastroenterol. 2020;26(37):5731–5744. doi: 10.3748/wjg.v26.i37.5731
- Pathak A, Agarwal N, Mandliya J, et al. Abernethy malformation: a case report. BMC Pediatrics. 2012;12(1):1. doi: 10.1186/1471-2431-12-57
- Duarte-Mesquita R, Sousa M, Vilaverde F, Cardoso R. Abernethy malformation : beware in cases of unexplained hepatic encephalopathy in adults. BJR| Case Reports. 2017;4(1):20170054. doi: 10.1259/bjrcr.20170054
- Allegritti M, Enrico B, Basile E, et al. Non-cirrhotic extra-hepatic porto-systemic shunt causing adult-onset encephalopathy treated with endovascular closure. Digestive Dis Sci. 2020;65(4):946–951. doi: 10.1007/s10620-019-06024-4
- Alvi AA, Pichardo J, Gupta S, et al. An interesting case of congenital intrahepatic porto-hepatic shunt as a cause of unexplained encephalopathy. Cureus. 2020;12(4):e7639. doi: 10.14309/01.ajg.0000598392.71372.f2
- De Vito C, Tyraskis A, Davenport M, et al. Histopathology of livers in patients with congenital portosystemic shunts (Abernethy malformation): A case series of 22 patients. Virchows Archiv. 2019;474(1):47–57. doi: 10.1007/s00428-018-2464-4
- Lautz TB, Shah SA, Superina RA. Hepatoblastoma in children with congenital portosystemic shunts. J Pediatric Gastroenterology Nutrition. 2016;62(4):542–545. doi: 10.1097/MPG.0000000000001012
- Correa C, Luengas JP, Howard SC, Veintemilla G. Hepatoblastoma and abernethy malformation type I: Case report. J Pediatric Hematology/Oncology. 2017;39(2):e79–e81. doi: 10.1097/MPH.0000000000000650
- Kwapisz L, Wells MM, Judaibi BA. Abernethy malformation: Congenital absence of the portal vein. Can J Gastroenterol Hepatol. 2014;28(11):587–588. doi: 10.1155/2014/675812
- Benedict M, Rodriguez-Davalos M, Emre S, et al. Congenital extrahepatic portosystemic shunt (abernethy malformation type Ib) with associated hepatocellular carcinoma: Case report and literature review. Pediatric Developmental Pathology. 2017;20(4):354–362. doi: 10.1177/1093526616686458
- Özden İ, Yavru A, Güllüoğlu M, et al. Transplantation for large liver tumors in the setting of abernethy malformation. Exp Clin Transplantation. 2017;15(Suppl 2):82–85. doi: 10.6002/ect.TOND16.L23
- Lin KY, Chen H, Yu L. Pulmonary arterial hypertension caused by congenital extrahepatic portocaval shunt: A case report. BMC Cardiovascular Disorders. 2019;19(1):1–5. doi: 10.1186/s12872-019-1124-1
- Osorio MJ, Bonow A, Bond GJ, et al. Abernethy malformation complicated by hepatopulmonary syndrome and a liver mass successfully treated by liver transplantation. Pediatric Transplantation. 2011;15(7):149–151. doi: 10.1111/j.1399-3046.2010.01337.x
- Baiges A, Turon F, Simón-Talero M, et al. Congenital extrahepatic portosystemic shunts (Abernethy malformation): An international observational study. Hepatology. 2020;71(2):658–669. doi: 10.1002/hep.30817
- Ponziani FR, Faccia M, Zocco MA, et al. Congenital extrahepatic portosystemic shunt: description of four cases and review of the literature. J Ultrasound. 2019;22:349–358. doi: 10.1007/s40477-018-0329-y
- Sheth R, Sivakumar K. The Abernethy malformation with inferior caval vein hypoplasia: A tailored technique for transcatheter closure and an insight into embryological perspective. Cardiology Young. 2018;28(9):1169–1171. doi: 10.1017/S1047951118000884
- Li H, Ma Z, Xie Y, Tian F. Recurrent hyperammonemia after abernethy malformation type 2 closure: A case report. Ann Hepatol. 2017;16(3):460–464. doi: 10.5604/01.3001.0009.8603
- Kanazawa H, Nosaka S, Miyazaki O, et al. The classification based on intrahepatic portal system for congenital portosystemic shunts. J Pediatric Sur. 2015;50(4):688–695. doi: 10.1016/j.jpedsurg.2015.01.009
Supplementary files

