







Combination of familial transthyretin amyloidosis and hyperlipoproteinemia(a) in a patient with spinal canal stenosis: a case report
- Authors: Nguyen T.L.1, Reznik E.V.2,3
-
Affiliations:
- 108 Military Central Hospital
- City Clinical Hospital No 31 named after academician G.M. Savelieva
- The Russian National Research Medical University named after N.I. Pirogov
- Issue: Vol 6, No 1 (2025)
- Pages: 166-177
- Section: Case reports
- Submitted: 10.10.2024
- Accepted: 06.12.2024
- Published: 25.03.2025
- URL: https://jdigitaldiagnostics.com/DD/article/view/636886
- DOI: https://doi.org/10.17816/DD636886
- ID: 636886

Cite item
Full Text

Abstract
Hereditary transthyretin amyloidosis is a rare, progressive, systemic autosomal dominant disorder characterized by the extracellular deposition of insoluble amyloid fibrils in the peripheral nervous system, heart, and other organs. Among the specific signs of this condition, symptomatic spinal canal stenosis is prominent. Lipoprotein(a) is an atherogenic lipoprotein, and increased plasma concentrations are a significant risk factor for cardiovascular and cerebrovascular diseases. Data regarding the relationship between transthyretin amyloidosis and lipoprotein(a) levels are limited.
This article presents a clinical case of a patient with arterial hypertension, with blood pressure elevated to 150/90 mmHg for 5 years. Following a COVID-19 infection between June 2, 2021, and June 25, 2021, the patient experienced a marked increase in blood pressure to 290/150 mmHg; sharp left-sided chest pain lasting 20–30 minutes unrelated to physical activity, which was relieved with medication; and pain in the cervical and thoracic spine. Despite antihypertensive therapy, the patient’s blood pressure stabilized at 110/70 mmHg. Further evaluation revealed dyslipidemia, with increased low-density lipoprotein cholesterol levels at 4.53 mmol/L and lipoprotein(a) at 1.46 g/L. Doppler ultrasound revealed atherosclerosis in the extracranial parts of the brachiocephalic arteries, with up to 20% stenosis of the right internal carotid artery. Echocardiography showed thickening of the left ventricular wall, interatrial septum, and mitral valve leaflets, although the ejection fraction remained preserved. Magnetic resonance imaging of the spine revealed cervical spinal canal stenosis (C5–C6). Genetic testing identified a nucleotide sequence variant in the transthyretin gene (Chr18: 29171879 G>A, p. Arg5His) in the heterozygous state in the patient and her blood relatives. Specific anti-amyloid therapy with tafamidis was considered, and hypolipidemic therapy was initiated.
In patients with symptomatic spinal canal stenosis and left ventricular wall thickening, even in the presence of hypertension, comprehensive evaluation is crucial for the timely diagnosis and adequate management of amyloid cardiomyopathy. Thus, we describe the first reported clinical case of the combination of familial transthyretin amyloidosis and hyperlipoproteinemia(a).
Full Text
АКТУАЛЬНОСТЬ
Транстиретиновый амилоидоз (ATTR-амилоидоз) — мультисистемное заболевание, вызванное отложением в органах амилоидных фибрилл, белком-предшественником которых является транстиретин. При наличии мутации в его гене он превращается из тетрамера в мономер, неправильно свёртывается и агрегирует с образованием амилоидных фибрилл. Для данного заболевания характерен аутосомно-доминантный тип наследования. В зависимости от типа мутации может клинически проявляться в виде семейной амилоидной полинейропатии или кардиомиопатии, или семейного лептоменингеального амилоидоза. Доступность генетических, биохимических и иммуногистохимических диагностических тестов предоставляет возможность выявлять ATTR-амилоидоз у пациентов во многих странах мира, однако до сих пор его верификация часто затруднена и несвоевременна. Данный факт обусловлен гетерогенностью клинической картины, неотличимой от полинейропатии и кардиомиопатии другой этиологии [1–3], необходимостью при некоторых типах амилоидоза выполнять инвазивные методы исследования, включая биопсию миокарда. Усовершенствованные диагностические подходы, наряду с таргетной терапией, могут замедлить прогрессирование заболевания и улучшить качество жизни пациентов.
Липопротеин (а) [ЛП(а)] — подкласс липопротеинов плазмы человека. Он по структуре и липидному составу похож на частицу липопротеина низкой плотности (ЛПНП). ЛП(а) также содержит аполипопротеин B, но в отличие от ЛПНП, он связан ковалентно через дисульфидную связь с аполипопротеином (а). Для измерения концентрации ЛП(а) необходимо выполнять отдельный анализ. Следует отметить, что его относят к категории атерогенных липопротеинов, поэтому повышение концентрации ЛП(а) в плазме крови считают значимым фактором риска сердечно-сосудистых и цереброваскулярных заболеваний, в частности инфаркта миокарда, ишемического инсульта, аортального стеноза и сердечной недостаточности [4].
P. Westermark [5] полагает, что аполипопротеины, а также аполипопротеин Е — важные компоненты амилоидных фибрилл в тканях при всех типах амилоида. Однако отсутствуют сведения о характере ассоциации ATTR-амилоидоза с ЛП(а) и его возможном клиническом значении.
Мы приводим описание клинического случая пациентки с верифицированным семейным ATTR-амилоидозом и гиперлипопротеин(а)емией.
ОПИСАНИЕ СЛУЧАЯ
Анамнез
Пациентка 50 лет на протяжении 5 лет страдала артериальной гипертензией с повышением артериального давления (АД) до 150/90 мм рт. ст. В анамнезе отсутствовали сведения об остром инфаркте миокарда, остром нарушении мозгового кровообращения, ожирении (рост 170 см, масса тела 75 кг, стабильная на протяжении последних 5 лет), сахарном диабете. Пациентка отрицала наличие вредных привычек — курение и злоупотребление алкоголем. В течение нескольких лет она наблюдалась у невролога по поводу эссенциального тремора и гинеколога в связи с наличием фиброзно-кистозной мастопатии, фиброаденомы правой молочной железы, дисфункции яичников перименопаузального периода, внутреннего эндометриоза и атрофического вульвовагинита.
После перенесённой (с 02.06.2021 по 25.06.2021) новой коронавирусной инфекции (COVID-19) стала отмечать повышение АД до 290/150 мм рт. ст., колющие боли в левой половине грудной клетки продолжительностью до 20–30 мин без связи с физической нагрузкой, купирующиеся после приёма препарата Корвалол® (мяты перечной листьев масло + Фенобарбитал + Этилбромизовалерианат, ОАО «Фармстандарт-Лексредства», Россия), а также боли в области шейного и грудного отделов позвоночника. В октябре 2021 г. пациентка обратилась к кардиологу.
Результаты физикального, лабораторного и инструментального исследования
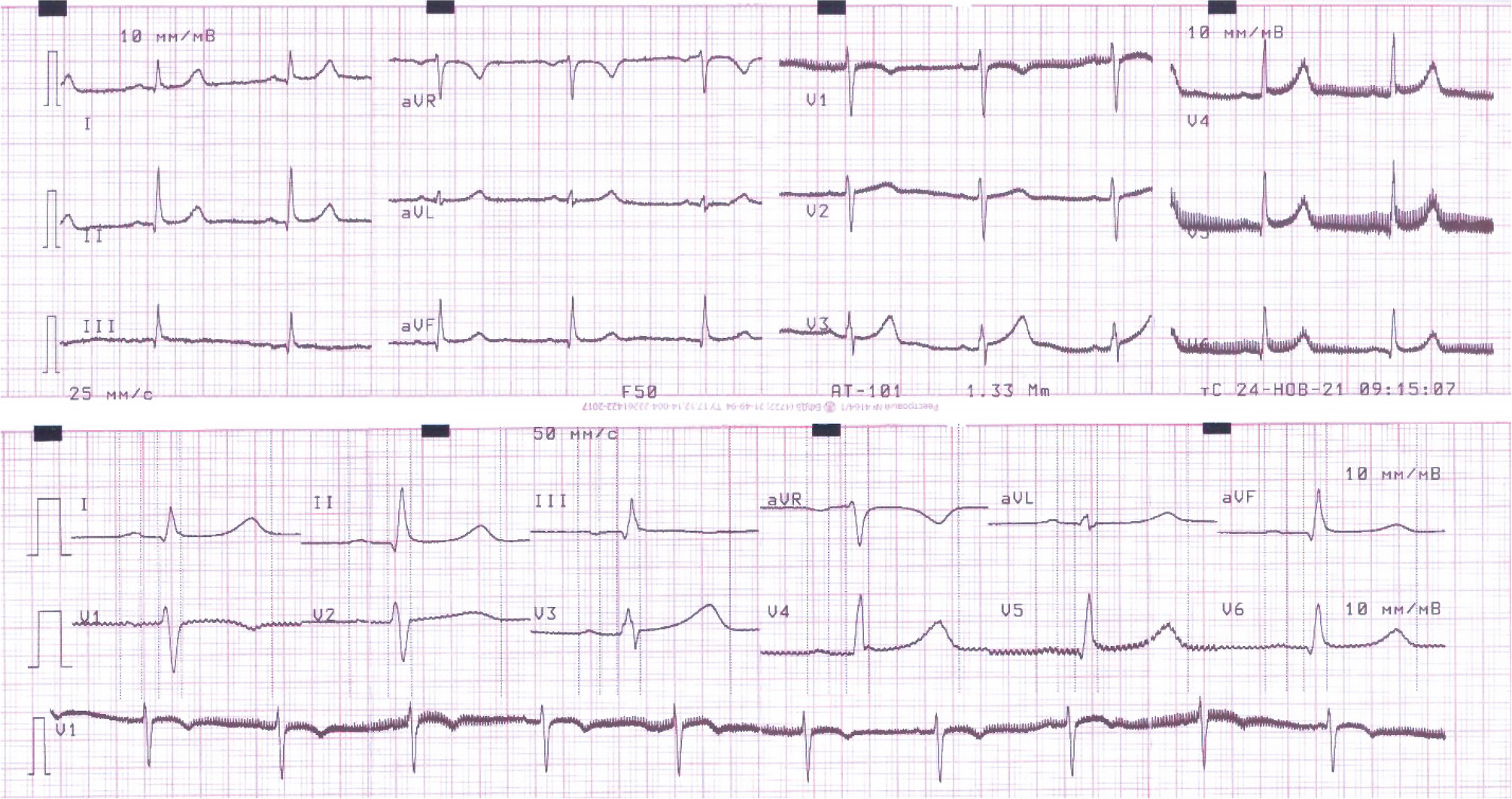
По данным электрокардиографии (ЭКГ) выявлено:
- синусовый ритм;
- частота сердечных сокращений (ЧСС) 64 в мин;
- нормальное направление электрической оси сердца;
- нормальный вольтаж комплекса QRS — выше 5 мм в отведениях от конечностей и выше 10 мм в прекардиальных;
- неспецифические изменения реполяризации в нижней стенке, боковой и верхушечной области левого желудочка (рис. 1).

Рис. 1. Электрокардиограмма в 12 отведениях пациентки с семейным транстиретиновым амилоидозом и гиперлипопротеин(а)емией.
При холтеровском (суточном) мониторировании сердечного ритма — основной ритм синусовый со средней ЧСС 78 в мин на протяжении дня, ночью — 73 в мин, 82 в мин за весь период регистрации. Максимальная ЧСС — 117 в мин, минимальная — 63 в мин, зафиксированы 4 желудочковые экстрасистолы, один наджелудочковый куплет. За время мониторирования пауз и диагностически значимой диспозиции сегмента ST не зарегистрировано.
Результаты общего и биохимического анализа крови пациентки представлены в табл. 1.
Таблица 1. Результаты общего и биохимического анализа крови пациентки
Показатели | Результат | Референсные значения |
Общий анализ крови | ||
Гемоглобин, г/л | 136 | 117–1601 |
Гематокрит | 0,38 | 0,35–0,471 |
Эритроциты, 1012/л | 4,1 | 3,8–5,31 |
Лейкоциты, 109/л | 4,99 | 4,5–11,32 |
Тромбоциты, 109/л | 177 | 180–320 |
Биохимический анализ крови | ||
Аспартатаминотрансфераза, МЕ/л | 17 | 5–34 |
Аланинаминотрансфераза, МЕ/л | 29 | 0–32 |
Общий белок, г/л | 73 | 65–85 |
Мочевина, ммоль/л | 3,7 | 2,5–8,33 |
Креатинин, мкмоль/л | 75 | 53–88 |
Натрий, ммоль/л | 143 | 130,5–156,6 |
Калий, ммоль/л | 4,2 | 3,44–5,30 |
Глюкоза, ммоль/л | 5,4 | 3,3–5,5 |
Холестерин липопротеинов низкой плотности, ммоль/л | 4,53 | до 3 |
Билирубин общий, мкмоль/л | 19 | 1,7–20,5 |
Липопротеин (а), г/л | 1,46 | до 0,5 |
Примечание. 1 — норма содержания эритроцитов у женщин в возрасте 45–64 лет; 2 — норма содержания лейкоцитов у женщин в возрасте 17–65 лет и старше. | ||
Скорость клубочковой фильтрации, рассчитанная с помощью формулы CKD-EPI, составила 80 мл/мин/1,73 м2. В общем анализе мочи отклонений не выявлено, однако концентрация микроальбумина в моче составила 4,1 мг/дл при референсном значении до 2 мг/дл.
При проведении ультразвуковой допплерографии сосудов выявлен атеросклероз внечерепных отделов брахицефальных артерий со стенозированием каротидной бифуркации и устья внутренней сонной артерии справа до 20%. Результаты эхокардиографии свидетельствовали о концентрической гипертрофии левого желудочка с толщиной межжелудочковой перегородки и задней стенки — 15,2 и 13,1 мм соответственно, тем не менее фракция выброса сохранена (55%). Также отмечены следующие изменения:
- диастолическая дисфункция по типу нарушения релаксации [I тип — максимальная скорость раннего диастолического наполнения левого желудочка (Е)=46 см/с; отношение Е к максимальной скорости потока во время предсердной систолы (Е/А)=0,6];
- утолщение межпредсердной перегородки до 7,4 мм и передней створки митрального клапана до 7 мм с её пролабированием в полость левого предсердия на 3,1 мм;
- митральная и трикуспидальная регургитация I степени (рис. 2).

Рис. 2. Результаты эхокардиография пациентки: a — утолщение межжелудочковой и межпредсердной перегородки (белые стрелки); b — утолщение передней створки митрального клапана (белая стрелка).
По результатам магнитно-резонансной томографии (МРТ) позвоночника выявлены протрузии дисков его шейного отдела (СIV–V, СV–VI, СVI–VII), стеноз позвоночного канала на уровне СV–VI, что считают «красным флагом» диагностики амилоидоза. Для исключения амилоидной этиологии стеноза позвоночного канала проведён генетический анализ сухих пятен крови пациентки, а затем её родственников. У пациентки, её родителей, двух сестёр и брата методом секвенирования по Сэнгеру выявлен вариант нуклеотидной последовательности в гене транстиретина (Chr18: 29171879 G>A, р.Arg5His) в гетерозиготном состоянии (рис. 3).

Рис. 3. Родословная пациентки с семейным транстиретиновым амилоидозом. Кружком обозначены женщины, квадратом — мужчины. Чёрным цветом показаны члены семьи с выявленной мутацией, белым — без выявленной мутации, серым — необследованные (обследование запланировано). Цифрами указан год рождения. ФП — фибрилляция предсердий в анамнезе у отца; ЛПа — повышение концентрации липопротеина (а), в частности у пациентки и её отца (у остальных членов семьи анализ на определение его содержания не проводили по техническим причинам).
В мае 2022 г. пациентке проведена сцинтиграфия миокарда с использованием радиофармпрепарата технеция (99mTc-пирофосфата), результаты которой не показали его поглощение, характерное для АТТR-амилоидоза (Grade 0).
Клинический диагноз
Основной диагноз: семейный ATTR-амилоидоз, нуклеотидный вариант с неопределённым клиническим значением в гетерозиготной состоянии в гене транстиретина.
Сопутствующие заболевания: гипертоническая болезнь III стадии. Целевой уровень АД не достигнут. Риск 4 (очень высокий) Дислипидемия (гиперлипопротеин(а)емия). Стеноз позвоночного канала на уровне СV–VI. Дорсопатия шейно-грудного и поясничного отделов позвоночника с хронической цервикокраниалгией (грыжа диска СV–VI, спондилоартроз, грыжа диска LIII–IV), затянувшееся обострение на фоне мышечнотонического синдрома. Эссенциальный тремор рук.
Лечение и рекомендации
Пациентке назначена трёхкомпонентная гипотензивная терапия и высокоинтенсивная терапия статином в комбинации с эзетимибом с достижением целевых значений АД и холестерина ЛПНП. С целью таргетной терапии гиперлипопротеин(а)емии ей предложено участие в клиническом рандомизированном двойном слепом плацебо-контролируемом многоцентровом исследовании (TQJ230). Тем не менее исследуемый препарат пелакарсен1 отменён в связи с аллергической реакцией в месте инъекции. Назначен инклисиран.
Пациентке рекомендовано динамическое наблюдение кардиолога и невролога, контроль АД, показателей липидного обмена, неврологических симптомов с регулярным рассмотрением вопроса о назначении специфической антиамилоидной терапии тафамидисом (рис. 4).

Рис. 4. Хронология заболеваний пациентки с семейным транстиретиновым амилоидозом и гиперлипопротеин(а)емией. ПЦР — полимеразная цепная реакция; ЛПНП — липопротеины низкой плотности; ЛП(а) — липопротеин (а); МРТ — магнитно-резонансная томография; ЭХО-КГ — эхокардиография; ЭНМГ — электронейромиография; РФП — радиофармацевтический препарат; ВКСП — вызванный кожный симпатический потенциал; С — шейный отдел позвоночника; пелакарсен — препарат не зарегистрирован на территории Российской Федерации.
Исход и результаты последующего наблюдения
Несмотря на назначенное лечение, в период с октября по декабрь 2023 г. пациентку дважды госпитализировали в связи с подозрением на острый коронарный синдром без подъёма сегмента ST на фоне повышения АД до 220/110 мм рт. ст., сопровождавшийся давящими болями за грудиной с иррадиацией в шею и под лопатку. Содержание тропонина I в крови <0,01 нг/мл, результаты ЭКГ не имели отрицательной динамики, коронароангиографию не проводили. При выполнении стресс-эхокардиографии после стабилизации показателей АД — субмаксимальная ЧСС достигнута, результат по критерию нарушенной локальной сократимости отрицательный.
При стимуляционной электронейромиографии, выполненной в мае 2024 г., выявлены незначительно выраженные аксональные изменения в правом малоберцовом нерве (преганглионарный уровень поражения), снижение амплитуды вызванного кожного симпатического потенциала с левой кисти и правой стопы.
Особенности семейного анамнеза
Следует отметить, что отец пациентки в течение длительного времени страдал артериальной гипертензией с повышением АД до 170/90 мм рт. ст., принимал телмисартан 40 мг/сут, на фоне чего стал отмечать склонность к гипотонии и самостоятельно перешёл на редкие приёмы данного препарата. В ноябре 2022 г. у него диагностировали постоянную форму фибрилляции предсердий. По данным ЭКГ выявлены низковольтажные комплексы QRS в отведениях от конечностей (рис. 5). Результаты биохимического анализа крови показали увеличение концентрации ЛП(а) до 0,67 г/л (до 0,52), креатинина до 107 мкмоль/л (53–8812), холестерина ЛПНП до 3,68 ммоль/л (до 312). При проведении иммунохроматографического анализа венозной крови отмечено увеличение содержания мозгового натрийуретического пропептида (NTproBNP) до 1881 пг/мл (0–1251). При оценке неврологического статуса пациента выявлен эссенциальный тремор, локальные нарушения проведения возбуждения по срединным нервам на уровне лучезапястных суставов (двусторонний синдром карпального канала). При проведении сцинтиграфии миокарда накопления радиофармпрепарата (99mTc-пирофосфата), характерного для АТТR-амилоидоза, не выявлено (Grade 0).

Рис. 5. Электрокардиограмма в 12 отведениях отца пациентки: a — скорость записи в стандартных и усиленных отведениях от конечностей 25 мм/с; b — скорость записи в отведениях с поверхности грудной клетки 50 мм/с. Стандартное напряжение на входе 1 мВ, что соответствует отклонению в 10 мм.
У матери пациентки, страдающей артериальной гипертензией и хронической сердечной недостаточностью также длительное время, по данным эхокардиографии выявлены следующие изменения:
- расширение аорты (диаметр восходящей аорты до 41 мм);
- снижение фракции выброса левого желудочка до 52%;
- утолщение межжелудочковой перегородки до 16,6 мм, задней стенки левого желудочка — до 13 мм, стенки правого желудочка — до 7 мм;
- умеренно выраженный кальциноз створок аортального и митрального клапана, а также их фиброзных колец.
Диастолическую функцию левого желудочка не оценивали. Кроме того, установлены изменения в биохимическом анализе крови:
- повышение концентрации креатинина до 107,6 мкмоль/л (53–8812 мкмоль/л) и холестерина ЛПНП до 5,36 ммоль/л (до 31 ммоль/л);
- увеличение активности сердечной фракции креатинфосфокиназы до 29,9 кЕд/л (0–2412 кЕд/л).
Брат пациентки не имел кардиологических симптомов и не принимал каких-либо лекарственных препаратов. Две сестры страдают артериальной гипертензией, у одной из них — нарушение функций щитовидной железы.
ОБСУЖДЕНИЕ
В данном клиническом случае у пациентки диагностировано сочетание семейного ATTR-амилоидоза и гиперлипопротеин(а)емии. Первое заболевание дебютировало с развития стеноза позвоночного канала и неврологической симптоматики, в то время как высокое содержание ЛП(а) выявлено при скрининговом обследовании по поводу дислипидемии и артериальной гипертензии. В поисковой системе PubMed мы не обнаружили публикаций, описывающих сочетание семейного ATTR-амилоидоза и гиперлипопротеин(а)емии.
Семейный ATTR-амилоидоз — редкое наследственное заболевание с аутосомно-доминантным типом наследования, проявляющееся обычно во взрослом возрасте полинейропатией и кардиомиопатией, характеризующееся прогрессирующим течением. ATTR-амилоидоз эндемичен для Северной Португалии, Швеции, Бразилии и Японии, где мутация Val30Met отмечена почти в 100% случаев [6, 7]. Россия не является эндемичным районом по АТТR-амилоидозу, однако частота выявления его постепенно увеличивается, а генетические мутации, обусловливающие данное заболевание, весьма разнообразны [6, 7].
ATTR-амилоидоз у пациентки заподозрен клинически на основании результатов эхокардиографии и признаков стеноза позвоночного канала. При проведении молекулярно-генетического анализа гена транстиретина (Chr18: 29171879 G>A) диагноз подтверждён (см. рис. 3). У пациентов с ATTR-амилоидозом стеноз позвоночного канала связан с отложением амилоида в жёлтой связке, особенно в поясничном отделе позвоночника. Многочисленные исследования рекомендуют проводить рутинный скрининг амилоидной кардиомиопатии у пациентов со стенозом позвоночного канала с целью выявления тех, кто нуждается в кардиологическом наблюдении. Отложение амилоида часто ассоциировано со стенозом позвоночного канала и его наличие может предоставить более раннюю возможность диагностировать системный амилоидоз [8].
Обычно с момента дебюта клинических проявлений до постановки диагноза в неэндемичных районах проходит 3–4 года. В нашем случае диагноз установлен в течение нескольких месяцев в связи с настороженностью клиницистов и наличием его «красных флагов». Выявленная мутация Chr18: 29171879 G>A (р.Arg5His) встречается крайне редко, с частотой аллеля 1,26/10 000. В регистре THAOS (The Transthyretin Amyloidosis Outcomes Survey) пациентов с ATTR-амилоидозом такой мутацией не было [6, 14, 33].
Важно отметить, что амилоидоз сердца следует заподозрить при утолщении стенки левого желудочка >12 мм и выявлении амилоида внесердечной локализации, даже при наличии артериальной гипертензии или других причин, вызывающих формирование его гипертрофии. При кардиологическом обследовании у описываемой пациентки выявлено значительное утолщение стенки миокарда левого желудочка (15,3 мм), межпредсердной перегородки (7,4 мм) и створки митрального клапана. Установлены единичные желудочковые экстрасистолы и наджелудочковый куплет. При проведении сцинтиграфии сердца не выявлено накопление изотопа в миокарде, что свидетельствует о диагностике заболевания на ранней стадии [3].
Проведение ЭКГ отцу пациентки позволило обнаружить низковольтажные комплексы QRS. Однако их отсутствие у пациентов молодого и среднего возраста не исключает диагноз амилоидной кардиомиопатии. Вероятно, вольтаж комплексов QRS отражает инфильтрацию амилоида в миокарде, выраженность которой неуклонно увеличивается с возрастом. При наличии у пациентов «красных флагов» амилоидоза, таких как стеноз позвоночного канала, двусторонний синдром карпального канала, нарушения ритма и проводимости неясного генеза, тремор рук и ног, полинейропатия неясного генеза, необходимо проводить скрининг на амилоидоз [9, 10]. Генетически верифицированный АТТR-амилоидоз у отца пациентки также сочетается с гиперлипопротеин(а)емией.
У матери пациентки выявлены признаки утолщения стенок сердца и дислипидемия, характеризующаяся увеличением холестерин ЛПНП до 5,36 ммоль/л. Однако анализ на определение содержания ЛП(а) в крови не проводили. В дальнейшем мы планируем его проведение у неё и других кровных родственников, а также — эхокардиографии, сцинтиграфии миокарда и электронейромиографии.
Несмотря на то что мы не проводили МРТ сердца, данный метод исследования является информативным в отношении амилоидной кардиомиопатии. Существует методика оценки позднего накопления гадолиния, учитывающая наличие циркулярных и/или трансмуральных его участков в базальных, средних и апикальных сегментах левого желудочка, а также наличие участков накопления в миокарде правого желудочка. Она позволяет провести дифференциальную диагностику амилоидоза лёгких цепей и его ATTR-типа с чувствительностью и специфичностью 82 и 76% соответственно [11]. При амилоидозе сердца удлинение нативного T1-времени релаксации наблюдают ещё до утолщения стенки левого желудочка, накопления контрастного вещества в миокарде и идентификации биомаркёров в крови. Проведение МРТ и оценка продольного (Т1) времени релаксации помогает отслеживать степень амилоидной инфильтрации сердца и изменения её с течением времени [12].
Возникает вопрос: когда следует начинать лечение пациентов с ATTR-амилоидозом, если у пациента выявлена мутация при генетическом тестировании или обнаружено отложение амилоида в биоптатах? Логичный ответ — как можно раньше, однако следует учитывать мнение доказательной медицины.
Согласно действующим рекомендациям по лечению ATTR-амилоидоза, которые отражают результаты клинических исследований в группах пациентов с кардиомиопатией и полинейропатией, необходимо выполнять сцинтиграфию с использованием радиофармпрепаратов. Обычно на этой стадии наблюдают картину сердечной недостаточности I–II функционального класса по классификации Нью-Йоркской кардиологической ассоциации (NYHA) [13, 14]. Она дебютирует, как правило, на III стадии инфильтративного заболевания сердца — необратимой [9]. Именно поэтому эффект от лечения может иметь ограничения. Данная пациентка и её родственники не соответствуют современным критериям начала антиамилоидной специфической терапии, несмотря на наличие и явное прогрессирование кардиомиопатии и полинейропатии. На наш взгляд, целесообразно проведение дополнительных клинических исследований среди таких пациентов с целью пересмотра сроков начала превентивной антиамилоидной терапии в сторону более ранних.
Гиперлипопротеин(а)емия является независимым фактором риска сердечно-сосудистых заболеваний, таких как инфаркт миокарда, инсульт, аортальный стеноз и сердечная недостаточность [4], поэтому анализ на определение концентрации ЛП(а) в крови необходимо проводить всем пациентам, минимум 1 раз в жизни. Исследования показали, что ЛП(а) способствует развитию сердечно-сосудистых заболеваний посредством проатерогенных, провоспалительных и антифибринолитических механизмов. Повышенный риск, связанный с ЛП(а), можно объяснить сочетанием высоких прокоагулянтных эффектов аполипопротеина (а) с атерогенными и провоспалительными эффектами аполипопротеин B-ассоциированными окисленными фосфолипидами [4]. У пациентки при дообследовании по поводу кардиальных жалоб выявлена дислипидемия, которая характеризовалась увеличением концентрации холестерина ЛПНП до 4,53 ммоль/л и ЛП(а) — до 1,46 г/л. Она сопровождалась атеросклерозом внечерепных отделов брахицефальных артерий со стенозированием каротидной бифуркации и устья внутренней сонной артерии справа. У её отца также выявлена дислипидемия, в частности гиперлипопротеин(а)емия.
С целью снижения риска сердечно-сосудистого события необходимо снижение концентрации ЛП(а). Статины неэффективны в лечении и могут, напротив, повышать его содержание [15]. В проспективном когортном исследовании Копенгагенского общего популяционного исследования выявлено, что при вторичной профилактике со средним периодом наблюдения 5 лет абсолютное снижение концентрации ЛП(а) на 50 и 99 мг/дл необходимо для уменьшения риска серьёзных сердечно-сосудистых событий на 20 и 40% соответственно [16]. В настоящее время проводят клинические исследования по лечению пациентов с гиперлипротеин(а)емией. В них используют несколько препаратов с положительными результатами, включая пелакарсен1 и инклисиран. Пелакарсен1 — антисмысловой олигонуклеотид, направленный на гепатоциты и матричную РНК гена ЛП(а). В рандомизированном контролируемом исследовании фазы 2 еженедельное или ежемесячное подкожное введение пелакарсена1 дозозависимо снижало концентрацию ЛП(a) на 35–80% [4]. Инклисиран — это новая терапия на основе малых интерферирующих РНК (антисмысловая). Связываясь с предшественником информационной РНК пропротеиновой конвертазы субтилизин-кексинового типа 9 (PCSK9), инклисиран ингибирует экспрессию её гена, что приводит к усилению рециркуляции гепатоцитов и мембранной экспрессии рецепторов ЛПНП, а также снижению концентрации ЛПНП и ЛП(а) до 30% [15]. Учитывая риск сердечно-сосудистых осложнений, мы назначили пациентке препарат, снижающий содержание ЛП(а). Сначала назначен пелакарсен1, однако из-за аллергической реакции после первой инъекции он отменён. В дальнейшем мы назначили инклисиран и сразу после первой инъекции в ноябре 2023 г. отмечено снижение концентрация ЛП(а) с 3,58 до 2,21 г/л и ЛПНП — с 2,21 до 1,03 ммоль/л (см. рис. 4). Эффективность лечения всё ещё требует дальнейшей оценки на последующих этапах лечения, а также с учётом клинических данных крупных исследований.
Аполипопротеины считают важными компонентами амилоидных фибрилл [5]. В амилоидных отложениях аполипопротеин E вместе с P компонентом и глюкозаминогликанами могут действовать как патологические молекулярные шапероны, которые вызывают β-складчатую конформацию в амилоидогенных полипептидах [5]. Аполипопротеин В в дополнение к аполипопротеину Е также обнаружен в амилоидных фибриллах в головном мозге пациентов с болезнью Альцгеймера [17]. При этом связанном с амилоидом расстройстве аллель аполипопротеина Е-4 используют в качестве генетического фактора риска [18]. Вариантные формы аполипопротеина А-I также связаны с семейным амилоидозом [19], характеризующимся отложением N-концевых фрагментов этого белка.
В исследованиях у пациентов с семейной амилоидной полинейропатией вследствие мутации Val30Met в гене транстиретина обнаружено, что он связан с липопротеинами [20, 21]. В частности, отмечено, что фракции липопротеина высокой плотности (ЛПВП) и ЛПНП содержат данный белок [20]. Транстиретин присутствовал во фракции ЛПВП в одинаковой степени как у здоровых людей, так и у пациентов с мутацией Val30Met, тогда как фракция ЛПНП имела большее содержание транстиретина у пациентов с мутацией Val30Met [20]. Однако связь между ЛП(а) и ATTR-амилоидозом до сих пор окончательно не ясна. Возникает потребность в дальнейшем изучении этой ассоциации и возможностей её клинического применения для профилактики и лечения данных заболеваний.
Следует также обсудить лечение артериальной гипертензии у данной пациентки. Взаимоотношения COVID-19 и артериальной гипертензии многообразны:
- сходство патогенеза с развитием системного воспаления и эндотелиальной дисфункции;
- активация ренин-ангиотензин-альдостероновой системы;
- возможное развитие артериальной гипертензии или её прогрессирование после перенесённой инфекции;
- дестабилизация АД как проявление постковидного синдрома [22].
В результате обследования 200 пациентов в Хорватии, перенёсших COVID-19, выявлено, что каждый седьмой подвержен риску развития новых случаев артериальной гипертензии или прогрессирования уже существующего заболевания [22]. Одним из проявлений постковидного синдрома при артериальной гипертензии, вероятно, является дестабилизация АД. Анализ данных регистра Евразийской ассоциации терапевтов выявил, что спустя 3 месяца после перенесённой COVID-19 у 20,1% обследованных зафиксирована неконтролируемая артериальная гипертензия [22]. Обнаружено периодическое повышение АД с его максимальным значением в первый месяц после заболевания, к третьему месяцу — его снижение, а к шестому — вторая волна повышения АД [22]. Почти 52% медицинских работников для контроля АД у пациентов с артериальной гипертензией, перенёсших COVID-19, перешли с монотерапии на двойную комбинированную, 20% — увеличили дозу двойной терапии и 13% — использовали тройную терапию [22]. Лечение артериальной гипертензии после COVID-19 — непростая задача. Несмотря на оптимальную антигипертензивную терапию, пациентку несколько раз госпитализировали в связи с подозрением на острый коронарный синдром без подъёма сегмента ST на фоне повышения АД до 220/110 мм рт. ст. (см. рис. 4).
ЗАКЛЮЧЕНИЕ
Таким образом, представлен случай семейного ATTR-амилоидоза, ассоциированного с редкой мутацией p. Arg123His, в сочетании с гиперлипопротеин(а)емией. Для своевременной диагностики и адекватной терапии данных патологических состояний необходима настороженность клиницистов в отношении амилоидоза и определение концентрации ЛП(а) у всех пациентов с сердечно-сосудистой патологией, в том числе с амилоидной кардиомиопатией. Внедрение современных подходов к диагностике и терапии редких заболеваний позволят улучшить качество и увеличить продолжительность жизни пациентов с изолированной и сочетанной патологией.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Источник финансирования. Авторы заявляют об отсутствии внешнего финансирования при проведении работы.
Раскрытие интересов. Авторы заявляют об отсутствии отношений, деятельности и интересов (личных, профессиональных или финансовых), связанных с третьими лицами (коммерческими, некоммерческими, частными), интересы которых могут быть затронуты содержанием статьи, а также иных отношений, деятельности и интересов за последние три года, о которых необходимо сообщить.
Вклад авторов. T.L. Nguyen — сбор и обработка материалов, анализ полученных данных, написание текста рукописи; Е.В. Резник — анализ полученных данных, редактирование текста рукописи. Все авторы одобрили рукопись (версию для публикации), а также согласились нести ответственность за все аспекты работы и гарантировали, что вопросы, связанные с точностью или добросовестностью любой части работы, будут должным образом рассмотрены и решены.
Информированное согласие на публикацию. Авторы получили письменное согласие пациентки на публикацию медицинских данных и фотографий в обезличенной форме в журнале Digital Diagnostics.
ADDITIONAL INFORMATION
Funding source. This article was not supported by any external sources of funding
Disclosure of interests. The authors declare that they have no relationships, activities or interests (personal, professional or financial) with third parties (commercial, non-commercial, private) whose interests may be affected by the content of the article, as well as no other relationships, activities or interests over the past three years that must be reported.
Authors' contribution. T.L. Nguyen: сollection and processing of materials, analysis of obtained data, writing the text of the manuscript; E.V. Reznik: analysis of obtained data, editing the manuscript. Thereby, all authors provided approval of the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Consent for publication. Written consent was obtained from the patient for publication of relevant medical information and all of accompanying images within the manuscript in Digital Diagnostics journal.
1 Препарат не зарегистрирован на территории Российской Федерации.
2 Референсные значения — диапазон значений, который считают нормальным для физиологических показателей у здоровых людей.
About the authors
Thanh L. Nguyen
108 Military Central Hospital
Author for correspondence.
Email: truongthianh0302@gmail.com
ORCID iD: 0000-0002-8856-4542
SPIN-code: 9408-1899
MD, Cand. Sci. (Medicine)
Viet Nam, HanoiElena V. Reznik
City Clinical Hospital No 31 named after academician G.M. Savelieva; The Russian National Research Medical University named after N.I. Pirogov
Email: elenaresnik@gmail.com
ORCID iD: 0000-0001-7479-418X
SPIN-code: 3494-9080
MD, Dr. Sci. (Medicine), Assistant Professor
Russian Federation, Moscow; MoscowReferences
- Reznik EV, Nguyen TL, Dikaeva MS, et al. Features of diagnostics and course of hypertrophic cardiomyopathy in real clinical practice. The Russian Archives of Internal Medicine. 2023;13(3):181–195. doi: 10.20514/2226-6704-2023-13-3-181-195 EDN: GGLHPG
- Reznik EV, Stepanova EA, Nguyen TL, et al. Retrospective analysis of cardiovascular involvement in patients with systemic amyloidosis. Cardiovascular Therapy and Prevention. 2021;20(1):35–46. doi: 10.15829/1728-8800-2021-2496 EDN: BYPVBU
- Reznik EV, Nguyen TL, Kudryavtseva MM, et al. Comparison of cardiac amyloidosis and hypertrophic cardiomyopathy: retrospective analysis of cardiac and kidney lesion. Russian Journal of Cardiology. 2023;28(11):5444. doi: 10.15829/15604071-2023-5444 EDN: JKLPYR
- Miksenas H, Januzzi JL, Natarajan P. Lipoprotein(a) and cardiovascular diseases. JAMA. 2021;326(4):352–353. doi: 10.1001/jama.2021.3632 EDN: OEOYNY
- Westermark P. The pathogenesis of amyloidosis: understanding general principles. Am J Pathol. 1998;152(5):1125–1127.
- Nikitin SS, Bardakov SN, Suponeva NA, et al. Phenotypic heterogeneity and diagnostic features of transthyretin amyloidosis with polyneuropathy. Neuromuscular Diseases. 2021;11(3):12–36. doi: 10.17650/2222-8721-2021-11-3-12-36 EDN: MSVKOX
- Rameev VV, Myasnikov RP, Vinogradov PP, et al. Systemic ATTR-amyloidosis, a rare form of internal organ damage. Rational Pharmacotherapy in Cardiology. 2019;15(3):349–358 doi: 10.20996/1819-6446-2019-15-3-349-358 EDN: VNIHZD
- Moore ZJ, Rizkalla JM, Weiner J, et al. Transthyretin amyloidosis in spinal canal stenosis: a systematic review. J Orthop. 2024;53:133–139. doi: 10.1016/j.jor.2024.02.047 EDN: NTUUWN
- Reznik EV, Nguyen TL, Ustyuzhanin DV, et al. Red flags to diagnose infiltrative cardiomyopathies. Russian Journal of Cardiology. 2023;28(1S):40–51. doi: 10.15829/1560-4071-2023-5259 EDN: ZGFWNJ
- Reznik EV, Nguyen TL, Stepanova EA, et al. Cardiac amyloidosis: internist and cardiologist insight. The Russian Archives of Internal Medicine. 2020;10(6):430–457. doi: 10.20514/2226-6704-2020-10-6-430-457 EDN: VHNDGN
- Dungu JN, Valencia O, Pinney JH, et al. CMR-based differentiation of AL and ATTR cardiac amyloidosis. JACC: Cardiovascular Imaging. 2014;7(2):133–142. doi: 10.1016/j.jcmg.2013.08.015
- Vergaro G, Aimo A, Barison A, et al. Keys to early diagnosis of cardiac amyloidosis: red flags from clinical, laboratory and imaging findings. European Journal of Preventive Cardiology. 2020;27(17):1806–1815. doi: 10.1177/2047487319877708 EDN: ACLJFW
- Vogel J, Carpinteiro A, Luedike P, et al. Current therapies and future horizons in cardiac amyloidosis treatment. Curr Heart Fail Rep. 2024;21(4):305–321. doi: 10.1007/s11897-024-00669-7 EDN: HFSLBF
- McDonagh TA, Gardner RS, Baumbach A, et al. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: developed by the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). With the special contribution of the heart failure association (HFA) of the ESC. European Journal of Heart Failure. 2022;24(1):4–131. doi: 10.1002/ejhf.2333 EDN: VGUJJF
- Chan DC, Watts GF. The promise of PCSK9 and lipoprotein(a) as targets for gene silencing therapies. Clinical Therapeutics. 2023;45(11):1034–1046. doi: 10.1016/j.clinthera.2023.07.008 EDN: QRJECJ
- Madsen CM, Kamstrup PR, Langsted A, et al. Lipoprotein(a)-lowering by 50 mg/dL (105 nmol/L) may be needed to reduce cardiovascular disease 20% in secondary prevention. Arteriosclerosis, Thrombosis, and Vascular Biology. 2020;40(1):255–266. doi: 10.1161/ATVBAHA.119.312951
- Handelmann G, Boyles J, Weisgraber K, et al. Effects of apolipoprotein E, beta-very low density lipoproteins, and cholesterol on the extension of neurites by rabbit dorsal root ganglion neurons in vitro. Journal of Lipid Research. 1992;33:1677–1688. doi: 10.1016/S0022-2275(20)41390-2
- Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis: An Official Journal of the American Heart Association. 1988;8(1):1–21. doi: 10.1161/01.ATV.8.1.1
- Genschel J, Haas R, Pröpsting MJ, et al. Apolipoprotein A-I induced amyloidosis. FEBS Letters. 1998;430(3):145–149. doi: 10.1016/S0014-5793(98)00668-1 EDN: PLCHLI
- Tanaka Y, Ando Y, Kumamoto T, et al. Changed affinity of apolipoprotein AII to high density lipoprotein (HDL) in patients with familial amyloidotic polyneuropathy (FAP) type I. Biochimica et Biophysica Acta (BBA) — Molecular Basis of Disease. 1994;1225(3):311–316. doi: 10.1016/0925-4439(94)90012-4
- Sousa MM, Berglund L, Saraiva MJ. Transthyretin in high density lipoproteins: association with apolipoprotein A-I. Journal of Lipid Research. 2000;41(1):58–65. doi: 10.1016/S0022-2275(20)32074-5
- Ryabova AY, Guzenko TN, Bykova AP, et al. Arterial hypertension and Covid-19: possible relationships. Modern Problems of Science and Education. 2023;(2):102. doi: 10.17513/spno.32438 EDN: VCQHPR
Supplementary files

