

")






Abernethy畸形:临床病例
- 作者: Panyukova A.V.1, Sinitsyn V.E.1, Mershina E.A.1, Rucheva N.A.2
-
隶属关系:
- Lomonosov Moscow State University, Medical Research and Educational Center
- V.I. Shumakov National Medical Research Center of Transplantology and Artificial Organs
- 期: 卷 4, 编号 2 (2023)
- 页面: 226-237
- 栏目: 临床病例及临床病例的系列
- ##submission.dateSubmitted##: 06.03.2023
- ##submission.dateAccepted##: 03.04.2023
- ##submission.datePublished##: 12.07.2023
- URL: https://jdigitaldiagnostics.com/DD/article/view/289714
- DOI: https://doi.org/10.17816/DD289714
- ID: 289714

如何引用文章

详细
先天性肝外门腔分流是与部分或全部门静脉血转入全身血液有关的罕见血管异常。先天性肝外门腔分流被称为Abernethy畸形。由于发病率低并临床表现多样,这种病理的识别是一个诊断问题。
本文描述了一个15岁患者的Ib型Abernethy畸形的临床病例,该患者长期有高血压、反复鼻出血、胸痛、头晕、呼吸困难、运动耐力低下、便血、上腹痛、恶心和瘙痒等病史。经过全面检查,患者被诊断为门静脉系统异常:门静脉导管扩张,直接流入下腔静脉。还发现了肝实质中的多个结节、扩张的心腔、心肌肥厚和肺动脉高压。鉴于症状的严重性以及分流的大小和类型,一个多学科科联合会诊建议进行肝移植。
我们在本文中讨论了门静脉系统异常的诊断算法和其他可能的治疗方案。
关键词
全文:
现实性
先天性门体分流(congenital portosystemic shunts,CPS)是与部分或全部门静脉血转入全身血液有关的罕见血管异常。据估计,CPS在新生儿中的发病率为1:30 000,在老年患者中的发病率为1:50 000[1]。由于血管解剖的巨大变异性,CPS的分类非常复杂。CPS可分为肝内分流和肝外分流,部分或完全无门静脉血流[2]。先天性肝外门腔分流被称为Abernethy畸形(John Abernethy),J.Abernethy于1793年首次描述了这一病理现象[3]。然而,先天性肝外门腔分流的病例报道很少。
临床病例描述
关于患者
一名15岁的男孩因长期上腹痛和恶心入院。患者还伴有发作性高血压(高达160/90 mm Hg)、反复鼻出血、胸痛、头晕、呼吸困难、运动耐量低、便血和长期皮肤瘙痒。
病史:入院前12年诊断出门脉高压(未提供医疗文件)。
实验和仪器诊断
肝功能检查显示了,丙氨酸氨基转移酶活性中度升高(59.8 U/L,正常值为13-50),天门冬氨酸氨基转移酶水平升高(67.1 U/L,正常值为15-46),γ-谷氨酰基转移酶(91 U/L,正常值为2-42)、碱性磷酸酶(316 U/L,正常值52-171 )、总胆红素(39.2 μmol/L,正常值3.4-17.1)、直接胆红素(12.5 μmol/L,正常值0-5 )水平升高,白蛋白浓度略有下 降(40.2 g/L,正常值41-55)。
血液常规检查和凝血检查都在正常范围内;血尿素氮和血清肌酐也在参考值范围内。
经胸超声心动图显示了心腔扩张、心肌肥 厚( 左心室壁厚为1.6 cm)、收缩期肺动脉高 压( 肺动脉收缩压为40 mm Hg)。观察到主动脉异位(纤维环水平的直径为3.4 cm,主动脉窦水平的为5.1 cm,升主动脉水平的为4.0 cm)。未发现左心室流出道狭窄和心室壁运动减弱;左心室功能保留。
腹部超声检查结果:肝脏增大,有很多结节;实质结构改变;纤维化的症状;肝门区域无明显门静脉主干或分支;肝脏血管形态变形;肝静脉狭窄。还发现门静脉高压和脾脏中度肿大。
为了评估肝结节,分析了血清甲胎蛋白肿瘤标志物水平:浓度正常(1.72 IU/ml)。
为了确诊和明确血管解剖结构,还进行了其他影像学检查。
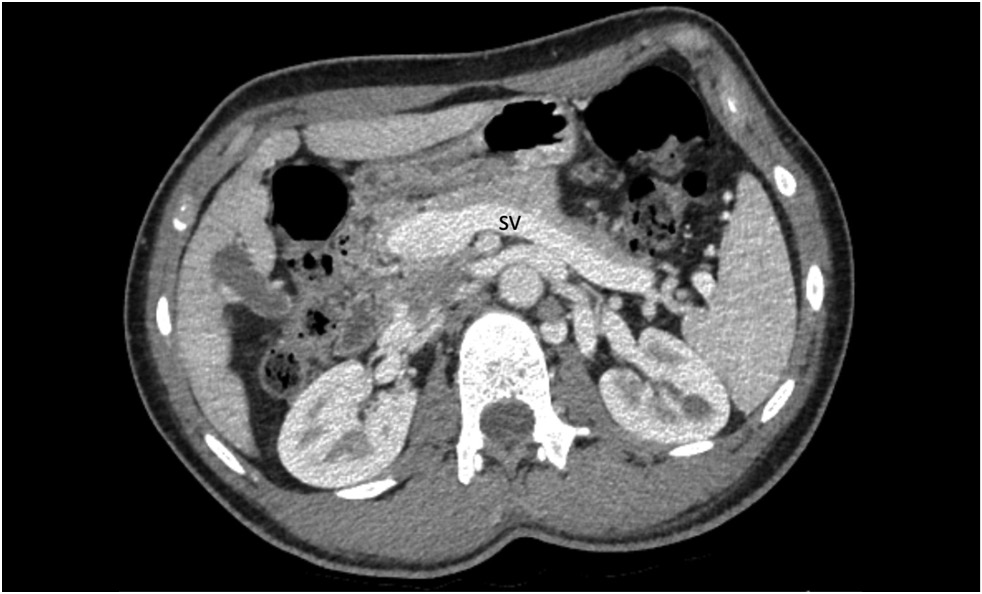
增强造影剂和多平面重建的腹部电子计算机断层扫描(CT)显示了,直径为12 mm的脾静 脉( 图1)和肠系膜上静脉合并在一起,形成直径达28 mm的门静脉导管(图2,图3)。门静脉导管绕过肝门直接流入下腔静脉(图4)。肝脏和脾脏中度肿大,肝实质轻度异型造影剂强化。根据检查结果,诊断为Abernethy畸形Ib型。

图1。造影剂计算机断层扫描,门静脉相,轴向投影:脾静脉(SV)扩张。

图2。造影剂计算机断层扫描,门静脉相,正面投影:脾静脉(SV)和肠系膜上静脉(SMV)合并形成门静脉导管(箭头)。

图3。造影剂计算机断层扫描,门静脉相,轴向投影:门静脉导管(箭头)。

图4。造影剂计算机断层扫描,门静脉相,正面投影:门静脉导管直接流入下腔静脉(箭头);肝脏增大;实质异质强化。
CT肺血管造影显示了,无异常血管分流,但证实了肺动脉干扩张(直径为40 mm)(图5)和心腔扩张,以及心肌肥厚(图6)。

图5。肺部计算机断层扫描血管造影,轴向投影:肺动脉干扩张。

图6。肺部计算机断层扫描血管造影,轴向投影:心肌肥厚。
治疗和预后
在与亚专科医生会诊后,由于保守治疗效果不佳、症状严重以及分流解剖结构,建议进行肝移植。目前,患者正在等待手术治疗。
讨论
发生机制
先天性门体分流和获得性门体分流的病因和发展有很大不同。先天性肝外门腔分流是由胎儿血管系统的异常形成或内陷发生的,而获得性分流是继发于肝脏疾病[2]。文献中关于先天性肝外门腔分流形成的理论主要有两种:先天性畸形和静脉导管异常。
门静脉系统的发育非常复杂,发生在胚胎第4周至第10周。总静脉系统由胚胎的前心静脉和后心静脉相互作用形成。门静脉系统由卵黄静脉形成,它们将血液从卵黄囊输送到静脉窦[4]。门静脉系统的发育中断导致先天性肝外门腔分流的发生。这种变异与合并先天性疾病密切相关。根据O.Bernard等人[1]研究结果,先天性心脏缺损是最常见的相关病变(265例中有45例)。其他报告的畸形包括肾脏、胆管(包括胆道闭锁)、消化系统、骨头和头脑的异常。
先天性肝外门腔分流发生的另一个机制是由于解剖缺陷或闭塞导致的胎儿静脉导管功能缺失。在正常发育的胎儿中,血液通过腔静脉从脐静脉流向下腔静脉,绕过肝脏。导管的功能性闭合是在出生后几分钟内自然发生的,而结构性闭合则是在大多数足月婴儿出生后的头几周发生的[5]。 脐静脉和静脉导管在出生后最初几个月解剖学上闭合,分别成为肝圆韧带和静脉韧带[4]。静脉导管的异常可以导致胎儿异常血管的形成。反过来,这些血管可能发展为异常分流。这导致门静脉系统发育不全。在某些先天性肝外门腔分流病例中,可发现无静脉导管[6,7]。
分类法
先天性肝外门腔分流的一个广泛使用的分类是由G.Morgan和R.Superina于1994年提出的分类系统(表1)。根据该分类法,Abernethy畸形根据肝内门脉系统的通畅程度分为两型[8]。I型为完全性门体分流,同时II型为部分血液分流至全身静脉,门静脉系统有一定程度的发育(图7)。根据先天性肝外门腔分流的类型,建议不同的治疗方案[9]。

图7。门静脉的正常解剖和分流的分类:a——门静脉的正常解剖;b——先天性肝外门腔分流,Ia型;c——先天性肝外门腔分流,Ib型;d——先天性肝外门腔分流,II型。
注:IVC(inferior vena cava)——下腔静脉;PV(portal vein)——门静脉;SV(splenic vein)——脾静脉;SMV(superior mesenteric vein)——肠系膜上静脉;Shunt——分流。
表1。根据G.Morgan和R.Superina[8]的门脉系统异常分类
I型 | 肝脏没有门静脉血液灌注——完全分流 |
Ia:肠系膜上静脉和脾静脉分别排入体循环 | |
Ib:肠系膜上静脉和脾静脉形成一条排入体循环的门静脉主干 | |
II型 | 肝脏有门静脉血液灌注——部分分流(如门静脉-肝静脉吻合术) |
IIa:先天性 | |
IIb: 获得性 |
临床表现和并发症
临床表现各不相同,取决于通过分流的血流量,其范围从无症状的成年患者的偶然发 现[10 ,11]到复杂先天性畸形[12]、严重低氧血症[13]、脑病[14,15]或肝肿瘤[16]。对于大多数患者来说,症状是非特异性的,如肝衰竭的急性失代或肝硬化。X.Lin等人[17]研究了703例先天性肝外门腔分流患者的数据。这些数据共发表在451篇文章中。他们的研究报告显示,大多数Abernethy畸形患者为儿童或18岁以下的年轻人。血流分流程度较高的严重先天性畸形的诊断年龄通常较小。如果存在部分血液分流,这种疾病在成年前可能没有症状。
半乳糖血症可能是先天性肝外门腔分流的首发症状。半乳糖血症是在新生儿早期的常规筛查中即可诊断的。半乳糖在肝脏中将半乳糖-1-磷酸尿苷转移酶代谢为葡萄糖。然而,在先天性肝外门腔分流患儿中,半乳糖会绕过肝脏。这导致其在全身血液中的水平升高[18,19]。一些研究者发现了,70%患有先天性肝外门腔分流的新生儿有高半乳糖血症[1]。新生儿早期可能出现的其他症状包括生长迟缓、新生儿胆汁淤积和肝性脑病[20]。
在病变较轻的患者中,先天性肝外门腔分流可能直到成年仍未被发现。表现可能与肝性脑病、肝肿瘤或肺动脉高压相关。
对于30%的先天性肝外门腔分流患者来说,可出现亚临床肝性脑病[21]。门静脉血液分流导致全身血液中的氨水平升高。氨在胃肠道的血液中形成。氨绕过肝脏直接进入下腔静脉。星形胶质细胞将氨代谢为谷氨酰胺。谷氨酰胺对头脑有毒性作用[14]。然而,高氨血症可能不伴有脑病,尤其是在年轻时。临床脑病在老年患者中更为常见,这可能是由于代偿能力较低所致[15]。由于症状的特异性较低,此类病例的诊断可能比较困难[14,22,23]。血清氨浓度升高而无肝硬化体征,应该进行进一步检查,是否存在肝外分流。
CPS患者容易发展为多发性肝肿瘤。关于CPS患者肝实质组织学变化的文献资料数量有限。C.De Vito等人[24]描述了涉及22例CPS患者的病例系列报道,其中19例先天性肝外门腔分流患者在他们的机构接受了长达15年的检查和随访。根据他们的研究结果,外周肝实质中最具有特征性的组织学发现包括门静脉明显薄壁道、动脉胆汁二联体、门静脉束和肝小叶中增大动脉轮廓的存在,以及在儿童中经常缺乏生理性的门静脉周围空泡化肝细胞。
先天性肝外门腔分流患者肝肿瘤的病理生理学仍不清楚。发生机制之一是由于肝脏再生能力下降。低门静脉血流导致将胰岛素和胰高血糖素到肝细胞的输送减少,从而增加了肝细胞损伤和肿瘤发生的风险[25]。此外,肝动脉血流增加可能与实质细胞的去分化有关[16]。
肝脏结节性病变是各种类型Abernethy畸形的典型表现。肝结节在大多数情况下是良性的,包括局灶性结节性增生、肝腺瘤和再生结节。大多数患者无症状。然而,对于其中一些患者来说,在腹腔发现肿块。在我们的病例中,肝结节是在腹部超声检查中意外发现的。
然而,并非所有肝脏肿块都是良性的。Abernethy畸形I型与肝母细胞瘤和肝细胞癌有关[26,27]。肝母细胞瘤很少见于先天性肝外门腔分流患儿。这些肿瘤预后不良。大多数病例都是致命的[25,26]。肝细胞癌多见于成人,但也有例外。因此,M.Benedict等人[28]发表了一例12个月大男孩的病例,他患有经组织学和免疫组化证实的肝细胞癌。肿瘤的诊断可能比较困难,因为有些病变的放射学特征不明确,可能被误认为良性肿块[11]。在这种情况下,通常需要进行活检。一种治疗方法是肝移植[29]。
一些先天性肝外门腔分流患者有肺动脉高压的体征,如呼吸困难[30,31]。严重的肺动脉高压可导致心源性晕厥,原因是前负荷降低和脑灌注不足[17]。在我们的病例中,肺动脉高压被确诊。
肝肺综合征发生于伴有门静脉高压的肝脏疾病患者。来自肠道的血管活性物质通过门体分流绕过肝循环,直接流入肺血管床;这导致血管扩张剂和血管收缩剂之间的不平衡,诱发肺动脉高压[9]。在这种情况下,肝血管异常是可以纠正的。
诊断和治疗
目前,还没有关于先天性肝外门腔分流诊断和治疗的公布指南。A.Baiges等人[32]提出了治疗先天性肝外门腔分流患者的模式(图8)。该模式基于包括66例患者的国际多中心研究结果。

图8。根据A.Baiges等人的研究,先天性肝外门腔分流患者的治疗模式[32]。
在我们的病例中,在进行腹部超声检查是怀疑为Abernethy畸形。通常,先天性肝外门腔分流的超声征象包括门静脉主干的缺失或发育不全、肝实质实性病灶、肝内门静脉血管和血流信号不足以及肝动脉肥大[33]。如果异常是超声检查时发现的,应该通过其他影像学方式,如CT或磁共振血管造影(MRA),进一步证实。增强CT可提供关于分流大小、方向和类型的重要信息。这有助于为每位患者选择最理想的治疗方法。此外,CT还可显示出和评估相关异常,包括肝脏肿块。MRA是显示出肝脏血管解剖的一种可靠和无创的方法。它不需要辐射,对软组织的对比度优于CT。此外,弥散加权图像和肝细胞特异性的造影剂可提供更多有价值的信息,以评估肝脏结节性病变和进一步作出决策。
治疗方法取决于分流的类型和大小、症状的严重程度、相关异常和并发症。无症状的患者可在医生指导下进行治疗。考虑到存在并发症的风险,L.Kwapisz等人[27]建议先天性肝外门腔分流患者接受常规临床检查、定期血液检查(包括肝功能检查)以及每年一次的肝脏影像学检查。
Abernethy畸形的治疗经验仍然是有限的。根据所描述的病例,目前的治疗方案包括介入或手术分流关闭术和肝移植。I型患者的长期治疗方案仅限于肝移植和手术前的支持治疗。对于II型先天性肝外门腔分流患者来说,根据并发症的发展和相关异常,有更多的治疗选择。其中之一是通过血管造影介入治疗(使用线圈或塞子)结扎或关闭门体分流[34]。但是,介入性关闭可能会导致复发性高氨血症,如之前的报道[35]。
使用球囊导管进行对分流的诊断性闭塞可能有助于评估两种类型先天性肝外门腔分流患者的肝内门脉系统[35]。该检查可显示出门静脉的小分支。超声检查不能显示出这些分支。H.Kanazawa等人[36],根据分流闭塞试验的结果,提出了肝内门脉系统的新分类(轻度、中度和重度)。肝内门脉系统分类与分流闭塞的门静脉压力、组织病理学检查结果、术后门静脉血流和肝脏再生相关。该分类有助于决定进行一期或二期分流关闭或肝移植。
结论
Abernethy畸形是一种罕见病。它与严重的并发症和不良预后有关。先天性肝外门腔分流的诊断具有挑战性,因为其发病率低、症状无特异性、不同器官系统是累及的以及表现中存在变异性。影像学在诊断和治疗计划中起着重要作用。早期发现和个体化治疗对预防并发症至关重要。需要对患者进行长期随访和恶性肿瘤监测。
ADDITIONAL INFORMATION
Funding source. This study was not supported by any external sources of funding.
Competing interests. The authors declare that they have no competing interests.
Authors’ contribution. All authors made a substantial contribution to the conception of the work, acquisition, analysis, interpretation of data for the work, drafting and revising the work, final approval of the version to be published and agree to be accountable for all aspects of the work.
Consent for publication. Written consent was obtained from the patient's parents for publication of relevant medical information and all of accompanying images within the manuscript in Digital Diagnostics journal.

作者简介
Alexandra V. Panyukova
Lomonosov Moscow State University, Medical Research and Educational Center
Email: panyukovaalexandra@gmail.com
ORCID iD: 0000-0002-5367-280X
俄罗斯联邦, Moscow
Valentin E. Sinitsyn
Lomonosov Moscow State University, Medical Research and Educational Center
Email: vsini@mail.ru
ORCID iD: 0000-0002-5649-2193
SPIN 代码: 8449-6590
MD, Dr. Sci. (Med); Professor
俄罗斯联邦, MoscowElena A. Mershina
Lomonosov Moscow State University, Medical Research and Educational Center
Email: elena_mershina@mail.ru
ORCID iD: 0000-0002-1266-4926
SPIN 代码: 6897-9641
MD, Cand. Sci. (Med.), Assistant Professor
俄罗斯联邦, MoscowNatalya A. Rucheva
V.I. Shumakov National Medical Research Center of Transplantology and Artificial Organs
编辑信件的主要联系方式.
Email: rna1969@yandex.ru
ORCID iD: 0000-0002-8063-4462
MD, Cand. Sci. (Med.)
俄罗斯联邦, Moscow参考
- Bernard O, Franchi-Abella S, Branchereau S, et al. Congenital portosystemic shunts in children: Recognition, evaluation, and management. Seminars Liver Dis. 2012;32(4):273–287. doi: 10.1055/s-0032-1329896
- Papamichail M, Pizanias M, Heaton N. Congenital portosystemic venous shunt. Eur J Pediatrics. 2018;177(3):285–294. doi: 10.1007/s00431-017-3058-x
- Abernethy J. Account of two instances of uncommon formation in the viscera of the human body: From the philosophical transactions of the royal society of London. Med Facts Observations. 1797;(7):100–108.
- Guérin F, Blanc T, Gauthier F, et al. Congenital portosystemic vascular malformations. Seminars Pediatric Sur. 2012;21(3):233–244. doi: 10.1053/j.sempedsurg.2012.05.006
- Born M. The ductus venosus. RoFo Fortschritte Gebiet Rontgenstrahlen Bildgebenden Verfahren. 2021;193(5):521–526. doi: 10.1055/a-1275-0984
- Baller SE, Reinehr M, Haslinger C, et al. Case report of neonatal ductus venosus atresia. J Neonatal-Perinatal Med. 2021;14(2):307–312. doi: 10.3233/NPM-190398
- Franchi-Abella S, Branchereau S, Lambert V, et al. Complications of congenital portosystemic shunts in children: Therapeutic options and outcomes. J Pediatric Gastroenterol Nutrition. 2010;51(3):322–330. doi: 10.1097/MPG.0b013e3181d9cb92
- Morgan G, Superina R. Congenital absence of the portal vein: Two cases and a proposed classification system for portasystemic vascular anomalies. J Pediatric Sur. 1994;29(9):1239–1241. doi: 10.1016/0022-3468(94)90812-5
- Tang H, Song P, Wang Z, et al. A basic understanding of congenital extrahepatic portosystemic shunt: Incidence, mechanism, complications, diagnosis, and treatment. Intractable Rare Dis Res. 2020;9(2):64–70. doi: 10.5582/irdr.2020.03005
- Păcurar D, Dijmărescu I, Dijmărescu AD, et al. A case report on an incidental discovery of congenital portosystemic shunt. Medicine. 2019;98(31):e16679. doi: 10.1097/MD.0000000000016679
- Shah A, Aziz A, Awwad A, et al. Incidental radiological diagnosis of asymptomatic Abernethy malformations: Two case reports. BJR|Case Reports. 2017;3(1):20150496. doi: 10.1259/bjrcr.20220059
- Yangín-Ergon E, Ermis N, Colak R, et al. Abernethy malformation type 2 and biliary atresia coexistence: A rare cause of infantile liver transplant. Euroasian J Hepatogastroenterol. 2018;8(2):163–166. doi: 10.5005/jp-journals-10018-1283
- Sahu MK, Bisoi AK, Chander NC, et al. Abernethy syndrome, a rare cause of hypoxemia: A case report. Ann Pediatric Cardiol. 2015;8(1):64–66. doi: 10.3389/fcvm.2021.784739
- Lux D, Naito A, Harikrishnan S. Congenital extrahepatic portosystemic shunt with progressive myelopathy and encephalopathy. Practical Neurol. 2019;19(4):368–371. doi: 10.1136/practneurol-2018-002111
- Merola E, Cao M, La Starza S. et al. Portosystemic encephalopathy in an 86-year-old patient: A clinical challenge. Acta Gastro-Enterologica Belgica. 2016;79(1):58–59.
- Sharma R, Suddle A, Quaglia A, et al. Congenital extrahepatic portosystemic shunt complicated by the development of hepatocellular carcinoma. Hepatobiliary Pancreatic Dis Int. 2015;14(5):552–557. doi: 10.1016/S1499-3872(15)60418-0
- Lin X, Rao J, Xiang Y, et al. Case report: A rare syncope case caused by abernethy II and a review of the literature. Frontiers Cardiovascular Med. 2022;8:2050. doi: 10.3389/fcvm.2021.784739
- Hasegawa T, Sato T, Ishii T, et al. Oral sodium phenylbutyrate for hyperammonemia associated with congenital portosystemic shunt: A case report. J Pediatric Endocrinol Metabolism. 2021;34(3):407–410. doi: 10.1515/jpem-2020-0603
- Peček J, Fister P, Homan M. Abernethy syndrome in Slovenian children: Five case reports and review of literature. World J Gastroenterol. 2020;26(37):5731–5744. doi: 10.3748/wjg.v26.i37.5731
- Pathak A, Agarwal N, Mandliya J, et al. Abernethy malformation: a case report. BMC Pediatrics. 2012;12(1):1. doi: 10.1186/1471-2431-12-57
- Duarte-Mesquita R, Sousa M, Vilaverde F, Cardoso R. Abernethy malformation : beware in cases of unexplained hepatic encephalopathy in adults. BJR| Case Reports. 2017;4(1):20170054. doi: 10.1259/bjrcr.20170054
- Allegritti M, Enrico B, Basile E, et al. Non-cirrhotic extra-hepatic porto-systemic shunt causing adult-onset encephalopathy treated with endovascular closure. Digestive Dis Sci. 2020;65(4):946–951. doi: 10.1007/s10620-019-06024-4
- Alvi AA, Pichardo J, Gupta S, et al. An interesting case of congenital intrahepatic porto-hepatic shunt as a cause of unexplained encephalopathy. Cureus. 2020;12(4):e7639. doi: 10.14309/01.ajg.0000598392.71372.f2
- De Vito C, Tyraskis A, Davenport M, et al. Histopathology of livers in patients with congenital portosystemic shunts (Abernethy malformation): A case series of 22 patients. Virchows Archiv. 2019;474(1):47–57. doi: 10.1007/s00428-018-2464-4
- Lautz TB, Shah SA, Superina RA. Hepatoblastoma in children with congenital portosystemic shunts. J Pediatric Gastroenterology Nutrition. 2016;62(4):542–545. doi: 10.1097/MPG.0000000000001012
- Correa C, Luengas JP, Howard SC, Veintemilla G. Hepatoblastoma and abernethy malformation type I: Case report. J Pediatric Hematology/Oncology. 2017;39(2):e79–e81. doi: 10.1097/MPH.0000000000000650
- Kwapisz L, Wells MM, Judaibi BA. Abernethy malformation: Congenital absence of the portal vein. Can J Gastroenterol Hepatol. 2014;28(11):587–588. doi: 10.1155/2014/675812
- Benedict M, Rodriguez-Davalos M, Emre S, et al. Congenital extrahepatic portosystemic shunt (abernethy malformation type Ib) with associated hepatocellular carcinoma: Case report and literature review. Pediatric Developmental Pathology. 2017;20(4):354–362. doi: 10.1177/1093526616686458
- Özden İ, Yavru A, Güllüoğlu M, et al. Transplantation for large liver tumors in the setting of abernethy malformation. Exp Clin Transplantation. 2017;15(Suppl 2):82–85. doi: 10.6002/ect.TOND16.L23
- Lin KY, Chen H, Yu L. Pulmonary arterial hypertension caused by congenital extrahepatic portocaval shunt: A case report. BMC Cardiovascular Disorders. 2019;19(1):1–5. doi: 10.1186/s12872-019-1124-1
- Osorio MJ, Bonow A, Bond GJ, et al. Abernethy malformation complicated by hepatopulmonary syndrome and a liver mass successfully treated by liver transplantation. Pediatric Transplantation. 2011;15(7):149–151. doi: 10.1111/j.1399-3046.2010.01337.x
- Baiges A, Turon F, Simón-Talero M, et al. Congenital extrahepatic portosystemic shunts (Abernethy malformation): An international observational study. Hepatology. 2020;71(2):658–669. doi: 10.1002/hep.30817
- Ponziani FR, Faccia M, Zocco MA, et al. Congenital extrahepatic portosystemic shunt: description of four cases and review of the literature. J Ultrasound. 2019;22:349–358. doi: 10.1007/s40477-018-0329-y
- Sheth R, Sivakumar K. The Abernethy malformation with inferior caval vein hypoplasia: A tailored technique for transcatheter closure and an insight into embryological perspective. Cardiology Young. 2018;28(9):1169–1171. doi: 10.1017/S1047951118000884
- Li H, Ma Z, Xie Y, Tian F. Recurrent hyperammonemia after abernethy malformation type 2 closure: A case report. Ann Hepatol. 2017;16(3):460–464. doi: 10.5604/01.3001.0009.8603
- Kanazawa H, Nosaka S, Miyazaki O, et al. The classification based on intrahepatic portal system for congenital portosystemic shunts. J Pediatric Sur. 2015;50(4):688–695. doi: 10.1016/j.jpedsurg.2015.01.009
补充文件




