

")








Мальформация Абернети: клинический случай
- Авторы: Панюкова А.В.1, Синицын В.Е.1, Мершина Е.А.1, Ручьёва Н.А.2
-
Учреждения:
- Московский государственный университет имени М.В. Ломоносова, Медицинский научно-образовательный центр
- Национальный медицинский исследовательский центр трансплантологии и искусственных органов имени академика В.И. Шумакова
- Выпуск: Том 4, № 2 (2023)
- Страницы: 226-237
- Раздел: Клинические случаи и серии клинических случаев
- Статья получена: 06.03.2023
- Статья одобрена: 03.04.2023
- Статья опубликована: 12.07.2023
- URL: https://jdigitaldiagnostics.com/DD/article/view/289714
- DOI: https://doi.org/10.17816/DD289714
- ID: 289714

Цитировать

Аннотация
В статье описан клинический случай мальформации Абернети типа Ib у 15-летнего пациента с длительным анамнезом повышенного артериального давления, рецидивирующими носовыми кровотечениями, болью в груди, головокружением, одышкой, низкой толерантностью к физической нагрузке, эпизодами крови в стуле, болью в эпигастральной области, тошнотой и зудом. В результате проведённого комплексного обследования у пациента была диагностирована аномалия развития портальной системы: расширенный кондуит воротной вены, впадающий непосредственно в нижнюю полую вену. Выявлены также множественные узлы в паренхиме печени, расширение камер сердца, гипертрофия миокарда и лёгочная гипертензия. Учитывая выраженность симптомов, размеры и тип шунта, междисциплинарным консилиумом рекомендована трансплантация печени.
В статье рассматриваются алгоритмы диагностики и другие возможные варианты лечения аномалий развития портальной системы.
Полный текст
АКТУАЛЬНОСТЬ
Врождённые портосистемные шунты (ВПШ) ― редкая врождённая аномалия, связанная с частичным или полным отведением портальной крови в системный кровоток. По оценкам, частота ВПШ составляет 1:30 000 новорождённых и 1:50 000 среди пациентов старшего возраста [1]. Классификация ВПШ сложна из-за значительной вариабельности анатомии сосудов. ВПШ подразделяются на внутри- и внепечёночные шунты с частичным или полным отсутствием портального кровотока [2]. Врождённые внепечёночные портосистемные шунты (ВВПШ) называют мальформацией Абернети (John Abernethy), который впервые описал патологию в 1793 году [3]. Однако случаев ВВПШ описано крайне мало.
ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ
О пациенте
Пятнадцатилетний юноша поступил в больницу для обследования по поводу хронической боли в эпигастральной области и тошноты. У больного также наблюдались эпизоды повышенного артериального давления (до 160/90 мм рт.ст.), повторяющиеся носовые кровотечения, боль в груди, головокружение, одышка, низкая переносимость физических нагрузок, гематохезия, продолжительный кожный зуд.
Анамнез болезни: за 12 лет до поступления диагностирована портальная гипертензия (медицинская документация не предоставлена).
Лабораторная и инструментальная диагностика
Функциональные пробы печени показали умеренное повышение активности аланиновой аминотрансферазы (59,8 Ед/л при норме 13–50), повышение уровней аспартатаминотрансферазы (67,1 Ед/л при норме 15–46), гамма-глутамилтрансферазы (91 Ед/л при норме 2–42), щелочной фосфатазы (316 Ед/л при норме 52–171), общего билирубина (39,2 мкмоль/л при норме 3,4–17,1), прямого билирубина (12,5 мкмоль/л при норме 0–5), а также незначительное снижение концентрации альбумина (40,2 г/л при норме 41–55).
Общий анализ крови и коагуляционные исследования были в пределах нормы; азот мочевины крови и сывороточный креатинин также соответствовали референсным значениям.
Трансторакальная эхокардиография выявила дилатацию камер сердца, гипертрофию миокарда (толщина стенок левого желудочка 1,6 см), систолическую лёгочную гипертензию (систолическое давление в лёгочной артерии 40 мм рт.ст.). Наблюдалась эктазия аорты (диаметр на уровне фиброзного кольца 3,4 см, синусов Вальсальвы ― 5,1 см, восходящей аорты ― 4,0 см). Стеноз выходного тракта левого желудочка и гипокинезия стенки желудочка не обнаружены; функция левого желудочка сохранена.
Результаты ультразвукового исследования (УЗИ) брюшной полости: увеличенная печень с множественными образованиями, размерами 7–8 мм; изменения в структуре паренхимы; признаки фиброза; отсутствие выраженного портального венозного ствола или ветвей в области ворот печени; деформированный, обеднённый сосудистый рисунок печени; стеноз печёночных вен. Отмечались также портальная гипертензия и умеренное увеличение селезёнки.
Для оценки потенциала злокачественности выявленных образований измерен уровень альфа-фетопротеина в сыворотке крови: концентрация соответствовала норме (1,72 МЕ/мл).
С целью подтверждения диагноза и уточнения анатомии сосудов проведены дополнительные визуализационные исследования.
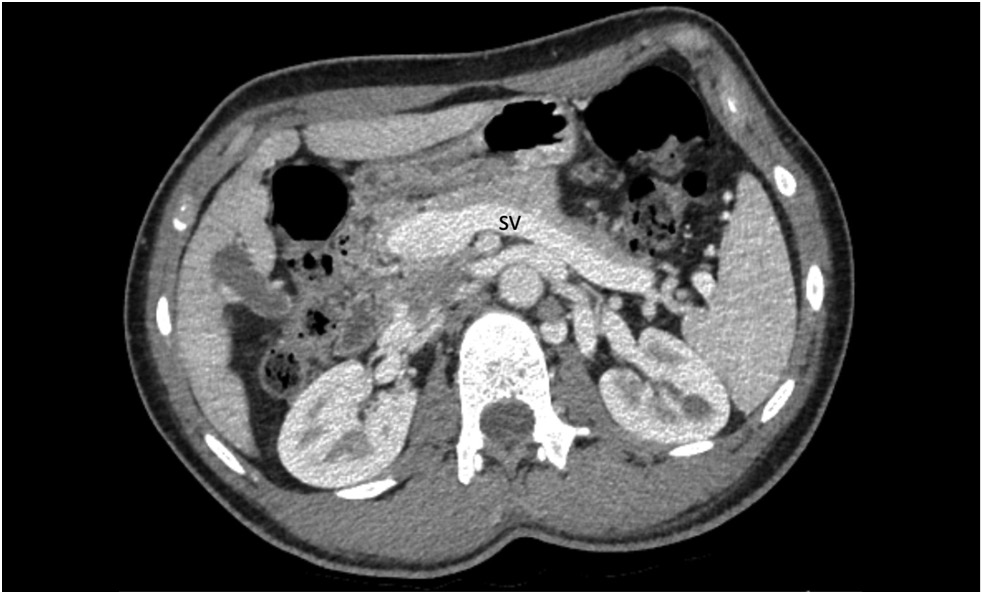
Компьютерная томография (КТ) брюшной полости с контрастным усилением и мультипланарной реконструкцией показала, что селезёночная вена диаметром 12 мм (рис. 1) и верхняя брыжеечная вена слились воедино, образовав кондуит воротной вены диаметром до 28 мм (рис. 2, 3), впадающий непосредственно в нижнюю полую вену, минуя ворота печени (рис. 4). Наблюдались также умеренное увеличение печени и селезёнки и слабое неоднородное контрастное усиление паренхимы печени. По результатам обследования диагностирована мальформация Абернети типа Ib.

Рис. 1. Компьютерная томография с контрастированием, портальная фаза, аксиальная проекция: расширенная селезёночная вена (SV).

Рис. 2. Компьютерная томография с контрастированием, портальная фаза, фронтальная проекция: селезёночная (SV) и верхняя брыжеечная (SMV) вены сливаются воедино, образуя кондуит воротной вены (стрелка).

Рис. 3. Компьютерная томография с контрастированием, портальная фаза, аксиальная проекция: кондуит воротной вены (стрелка).

Рис. 4. Компьютерная томография с контрастированием, портальная фаза, фронтальная проекция: кондуит воротной вены впадает непосредственно в нижнюю полую вену (стрелка); увеличение размеров печени; неоднородное контрастирование паренхимы печени.
КТ-ангиография лёгких не выявила аномальных сосудистых шунтов, однако подтвердила дилатацию лёгочного ствола (диаметр 40 мм) (рис. 5) и камер сердца, а также гипертрофию миокарда (рис. 6).

Рис. 5. Компьютерно-томографическая ангиография лёгких, аксиальная проекция: дилатация лёгочного ствола.

Рис. 6. Компьютерно-томографическая ангиография лёгких, аксиальная проекция: гипертрофия миокарда.
Лечение и прогноз
В связи с низкой эффективностью консервативного лечения, тяжестью симптомов и анатомией шунта на междисциплинарном консилиуме было рекомендовано проведение трансплантации печени. В настоящее время пациент ожидает хирургического вмешательства.
ОБСУЖДЕНИЕ
Механизмы возникновения
Этиология и развитие врождённых и приобретённых портосистемных шунтов существенно различаются. ВВПШ возникают из-за аномального формирования или инволюции сосудистой системы плода, тогда как приобретённые шунты вторичны по отношению к заболеваниям печени [2]. В литературе представлены две доминирующие теории формирования ВВПШ: врождённые пороки развития и аномалии венозного протока.
Развитие портальной системы — многоэтапный процесс, происходящий между 4-й и 10-й неделями внутриутробного развития. В отличие от полых вен, образующихся вследствие взаимодействия передней и задней кардиальных вен, портальная венозная система формируется из желточных вен, несущих кровь от желточного мешка к венозному синусу [4]. При нарушении развития портальной системы возникают ВВПШ. Этот вариант тесно связан с комбинированными врождёнными патологиями. По данным исследования O. Bernard и соавт. [1], врождённый порок сердца ― наиболее часто встречающаяся сопутствующая патология (45 из 265 случаев). В числе других зарегистрированных пороков развития отмечались аномалии головного мозга, почек, желчных протоков, включая билиарную атрезию, пищеварительной системы, костей.
Другой обсуждаемый механизм возникновения ВВПШ ― отсутствие функционирующего венозного протока у плода вследствие анатомических дефектов или окклюзии. У здорового плода кровь по венозному протоку поступает из пупочной вены в нижнюю полую вену, минуя печень. Функциональное закрытие венозного протока происходит в течение первых минут после рождения ребёнка, а структурное ― в течение первых недель жизни у большинства доношенных новорождённых [5]. Пупочная вена и венозный проток анатомически закрываются в первые месяцы жизни и становятся круглой связкой печени и венозной связкой соответственно [4]. Патологии венозного протока могут провоцировать образование аномальных сосудов у плода, которые, в свою очередь, могут превращаться в аномальные шунты, что приводит к гипоплазии портальной венозной системы. В некоторых описанных случаях ВВПШ отмечалось отсутствие венозного протока [6, 7].
Классификация
Широко используемая классификация ВВПШ ― это система классификации, предложенная G. Morgan и R. Superina в 1994 году (табл. 1), согласно которой мальформация Абернети делится на два типа в зависимости от наличия внутрипечёночной портальной системы [8]. Тип I определяется как полный портосистемный шунт, тогда как тип II описывается как частичное шунтирование крови в системные вены с некоторой степенью развития портальной системы (рис. 7). В зависимости от типа ВВПШ применяются различные терапевтические подходы [9].
Таблица 1. Классификация портосистемных аномалий по G. Morgan и R. Superina [8]
Тип I | Печень не перфузируется портальной кровью ― полный шунт |
Ia: верхняя брыжеечная вена и селезёночная вена дренируются отдельно в системный кровоток | |
Ib: верхняя брыжеечная вена и селезёночная вена формируют единый ствол воротной вены, который дренируется в системный кровоток | |
Тип II | Печень перфузируется портальной кровью ― частичный шунт (например, портально-печёночные венозные анастомозы) |
IIa: врождённый | |
IIb: приобретённый |

Рис. 7. Нормальная анатомия воротной вены и классификация шунтов: а ― нормальная анатомия воротной вены; b ― врождённый внепечёночный портосистемный шунт, тип Ia; c ― врождённый внепечёночный портосистемный шунт, тип Ib, d ― врождённый внепечёночный портосистемный шунт, тип II.
Примечание. IVC (inferior vena cava) ― нижняя полая вена; PV (portal vein) ― воротная вена; SV (splenic vein) ― селезёночная вена; SMV (superior mesenteric vein) ― верхняя брыжеечная вена; Shunt ― шунт.
Клинические проявления и осложнения
Описываемая патология отличается многообразием клинических проявлений. Выраженность симптомов зависит от объёма крови, протекающей через шунт, и варьирует от случайных находок у бессимптомных взрослых пациентов [10, 11] до сложных врождённых пороков развития [12], тяжёлой гипоксемии [13], энцефалопатии [14, 15] и злокачественных новообразований печени [16]. У большинства пациентов наблюдаются неспецифические симптомы, обусловленные острой декомпенсацией печёночной недостаточности или циррозом печени. В исследовании X. Lin и соавт. [17], основанном на анализе 451 статьи и включающем данные 703 пациентов с ВВПШ, сообщается, что большинство пациентов с мальформацией Абернети были детьми или молодыми людьми в возрасте до 18 лет. Тяжёлые врождённые патологии с высокой степенью шунтирования крови чаще диагностируются в более молодом возрасте. У пациентов с частичным шунтированием крови заболевание может протекать бессимптомно вплоть до зрелого возраста.
Первым признаком ВВПШ может быть галактоземия, диагностированная в раннем неонатальном периоде во время рутинного скрининга. Галактоза метаболизируется в печени ферментом галактозо-1-фосфат-уридилтрансфераза до глюкозы, однако у детей с ВВПШ галактоза с током крови минует печень, что приводит к повышению её уровня в системном кровотоке [18, 19]. По данным ряда исследователей, гипергалактоземия выявляется у 70% новорождённых с ВВПШ [1]. Другие возможные проявления в раннем неонатальном периоде ― задержка роста, неонатальный холестаз, печёночная энцефалопатия [20].
У пациентов с более лёгкой патологией ВВПШ может оставаться незамеченным до зрелого возраста. Проявления могут быть обусловлены симптомами, связанными с развитием печёночной энцефалопатии, новообразований в печени или лёгочной гипертензии.
У 30% пациентов с ВВПШ наблюдается субклиническая печёночная энцефалопатия [21]. Шунтирование портальной крови вызывает повышение уровня аммония в системном кровотоке. Аммиак, образуемый в желудочно-кишечном тракте, с током крови по шунту минует печень и поступает непосредственно в нижнюю полую вену. Астроциты метаболизируют аммоний до глутамина, который оказывает токсическое воздействие на головной мозг [14]. Однако гипераммониемия не всегда сопровождается энцефалопатией, особенно в молодом возрасте. Клиническая энцефалопатия чаще встречается у более взрослых пациентов, вероятно, вследствие снижения компенсаторных возможностей [15]. Диагностика в таких случаях может быть затруднена из-за низкой специфичности симптомов [14, 22, 23]. При выявлении повышенной концентрации аммиака в сыворотке крови у пациентов без признаков цирроза печени может быть целесообразно проведение дальнейшего обследования для исключения ВВПШ.
Пациенты с ВПШ подвержены к развитию множественных образований печени. Литературные данные о характере гистологических изменений паренхимы печени у пациентов с ВПШ ограничены. C. De Vito и соавт. [24] описали серию случаев с участием 22 пациентов с ВПШ, включая 19 пациентов с ВВПШ, которые были обследованы и наблюдались в их учреждении в течение 15 лет. Согласно полученным результатам, наиболее характерные гистологические находки в периферических участках паренхимы печени включали наличие портальных тонкостенных каналов, артериально-билиарных диад, увеличенных артериальных профилей в портальных трактах и дольках, а также частое отсутствие перипортально-вакуолизированных гепатоцитов у детей.
Патофизиология развития образований печени у пациентов с ВВПШ остаётся неясной. Один из возможных механизмов — снижение способности печени к регенерации. Низкий портальный кровоток приводит к снижению доставки инсулина и глюкагона к гепатоцитам, что увеличивает риск их повреждения и развития новообразований [25]. Кроме того, увеличение печёночного артериального кровотока может быть связано с дедифференцировкой паренхимальных клеток [16].
Частое проявление ВВПШ — узловые образования печени, выявляемые у пациентов с различными типами мальформации Абернети. В большинстве случаев данные новообразования имеют доброкачественную природу и являются очагами узловой гиперплазии, аденомами печени или узлами регенерации. Доброкачественные новообразования печени чаще протекают бессимптомно. В описываемом клиническом случае узловые образования также были случайной находкой во время УЗИ брюшной полости.
Однако не все выявляемые образования печени у пациентов с ВВПШ являются доброкачественными. Мальформация Абернети I типа связана с развитием гепатобластом и гепатоцеллюлярных карцином [26, 27]. Гепатобластомы — редкие низкодифференцированные опухоли печени, встречающиеся у детей с ВВПШ. Гепатобластома имеет крайне неблагоприятный прогноз, большинство описанных случаев у пациентов с ВВПШ были летальными [25, 26]. Гепатоцеллюлярные карциномы чаще наблюдаются во взрослом возрасте, но бывают исключения. Так, M. Benedict и соавт. [28] опубликовали случай 12-месячного мальчика с ВВПШ с гистологически и иммуногистохимически подтверждённой гепатоцеллюлярной карциномой. Диагностика злокачественных опухолей может быть затруднена, поскольку некоторые опухоли имеют неоднозначные визуальные характеристики и могут быть приняты за доброкачественные образования [11]. В таких случаях для постановки диагноза требуется биопсия. Один из вариантов лечения ― трансплантация печени [29].
У некоторых пациентов с ВВПШ наблюдаются симптомы лёгочной гипертензии: затруднённое дыхание и одышка [30, 31]. Тяжёлая лёгочная гипертензия может привести к кардиогенному обмороку вследствие снижения преднагрузки и низкой церебральной перфузии [17]. В описываемом клиническом случае также была диагностирована лёгочная гипертензия.
У пациентов с заболеваниями печени, сопровождающимися портальной гипертензией, может развиваться гепатопульмональный синдром. Вазоактивные медиаторы из кишечника, минуя печёночное кровообращение через портосистемный шунт, поступают непосредственно в лёгочное сосудистое русло, вызывая дисбаланс между вазодилатационными и вазоконстрикционными веществами, индуцируя лёгочную гипертензию [9]. Терапия данного состояния — коррекция сосудистых аномалий печени.
Диагностика и лечение
В настоящее время нет опубликованных клинических рекомендаций по диагностике и лечению ВВПШ. A. Baiges и соавт. [32] на основании результатов многоцентрового международного исследования, включавшего 66 пациентов, предложили свой алгоритм ведения пациентов с ВВПШ (рис. 8).

Рис. 8. Алгоритм ведения пациентов с врождённым внепечёночным портосистемным шунтом по A. Baiges и соавт. [32].
В описываемом клиническом случае подозрение на мальформацию Абернети возникло при проведении УЗИ брюшной полости. Как правило, ультразвуковые признаки ВВПШ включают отсутствие или гипоплазию воротной вены, очаговые образования в паренхиме печени, недостаточность внутрипечёночных портальных сосудов и сигналов потока, гипертрофию печёночной артерии [33]. Аномалии, выявленные в ходе УЗИ, должны быть дополнительно подтверждены с помощью других методов визуализации, таких как КТ или магнитно-резонансная ангиография (МР-ангиография). КТ с контрастным усилением даёт важную информацию о размере, расположении и типе шунта, что помогает в выборе наиболее оптимального подхода к лечению индивидуально для каждого пациента. Кроме того, КТ позволяет визуализировать и оценивать сопутствующие аномалии, включая образования в печени. МР-ангиография ― надёжный неинвазивный метод визуализации анатомии печёночных сосудов, который в отличие от КТ не сопряжён с рентгеновским излучением и имеет более высокий тканевой контраст. Кроме того, применение диффузионно-взвешенных изображений и гепатотропных контрастных препаратов может дать дополнительную ценную информацию для оценки узловых образований печени и последующего принятия решений о тактике ведения пациента.
Терапевтический подход зависит от типа и размеров шунта, тяжести симптомов, сопутствующих аномалий и осложнений. Бессимптомные пациенты могут находиться под медицинским наблюдением. Так, учитывая риск развития осложнений, L. Kwapisz и соавт. [27] рекомендуют пациентам с ВВПШ проходить плановые медицинские обследования, регулярные анализы крови, включая печёночные пробы, а также ежегодные визуализирующие исследования печени.
Опыт лечения пациентов с мальформацией Абернети по-прежнему невелик. Исходя из описанных случаев, современные варианты хирургического лечения включают интервенционное или хирургическое закрытие шунта и трансплантацию печени. Варианты долгосрочного лечения при I типе ограничиваются пересадкой печени с поддерживающей терапией в период ожидания операции. У пациентов с ВВПШ II типа имеется больший выбор вариантов терапии в зависимости от развившихся осложнений и сопутствующих аномалий. Один из них ― лигирование или закрытие портосистемного шунта с помощью интервенционной ангиографии [34]. Однако стоит учитывать, что интервенционное закрытие может вызвать рецидивирующую гипераммониемию [35].
Может оказаться полезным проведение диагностической окклюзии шунта баллонным катетером для оценки внутрипечёночной портальной системы у пациентов с обоими типами ВВПШ [35]. Этот тест позволяет визуализировать мелкие ветви воротной вены, которые невозможно увидеть на УЗИ. H. Kanazawa и соавт. [36] предложили новую классификацию внутрипечёночной портальной системы (лёгкая, умеренная и тяжёлая), исходя из результатов теста на окклюзию шунта. Классификация внутрипечёночной портальной системы коррелирует с портальным венозным давлением при окклюзии шунта, результатами гистопатологических исследований, послеоперационным портальным венозным кровотоком и регенерацией печени и полезна для принятия решения о проведении одно- или двухэтапного закрытия шунта или трансплантации печени.
ЗАКЛЮЧЕНИЕ
Мальформация Абернети ― редкая патология, связанная с тяжёлыми осложнениями и неблагоприятными исходами. Постановка диагноза ВВПШ представляет собой сложную диагностическую задачу из-за низкой распространённости, неспецифичности симптомов, вовлечения различных систем органов и вариабельности клинических проявлений. Методы медицинской визуализации играют важную роль в диагностике данной патологии и планировании лечения. Раннее выявление и индивидуальный подход к лечению пациента имеют решающее значение для предотвращения развития осложнений. Кроме того, требуются длительное медицинское наблюдение за пациентами и регулярные обследования для своевременного выявления злокачественных новообразований.
ДОПОЛНИТЕЛЬНО
Источник финансирования. Авторы заявляют об отсутствии внешнего финансирования при проведении поисково-аналитической работы.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Вклад авторов. Все авторы подтверждают соответствие своего авторства международным критериям ICMJE (все авторы внесли существенный вклад в разработку концепции, проведение поисково-аналитической работы и подготовку статьи, прочли и одобрили финальную версию перед публикацией).
Информированное согласие на публикацию. Законные представители пациента добровольно подписали информированное согласие на публикацию персональной медицинской информации в обезличенной форме в журнале Digital Diagnostics.
ADDITIONAL INFORMATION
Funding source. This study was not supported by any external sources of funding.
Competing interests. The authors declare that they have no competing interests.
Authors’ contribution. All authors made a substantial contribution to the conception of the work, acquisition, analysis, interpretation of data for the work, drafting and revising the work, final approval of the version to be published and agree to be accountable for all aspects of the work.
Consent for publication. Written consent was obtained from the patient's parents for publication of relevant medical information and all of accompanying images within the manuscript in Digital Diagnostics journal.
Об авторах
Александра Вадимовна Панюкова
Московский государственный университет имени М.В. Ломоносова, Медицинский научно-образовательный центр
Email: panyukovaalexandra@gmail.com
ORCID iD: 0000-0002-5367-280X
Россия, Москва
Валентин Евгеньевич Синицын
Московский государственный университет имени М.В. Ломоносова, Медицинский научно-образовательный центр
Email: vsini@mail.ru
ORCID iD: 0000-0002-5649-2193
SPIN-код: 8449-6590
д-р мед. наук, профессор
Россия, МоскваЕлена Александровна Мершина
Московский государственный университет имени М.В. Ломоносова, Медицинский научно-образовательный центр
Email: elena_mershina@mail.ru
ORCID iD: 0000-0002-1266-4926
SPIN-код: 6897-9641
канд. мед. наук, доцент
Россия, МоскваНаталья Александровна Ручьёва
Национальный медицинский исследовательский центр трансплантологии и искусственных органов имени академика В.И. Шумакова
Автор, ответственный за переписку.
Email: rna1969@yandex.ru
ORCID iD: 0000-0002-8063-4462
канд. мед. наук
Россия, МоскваСписок литературы
- Bernard O., Franchi-Abella S., Branchereau S., et al. Congenital portosystemic shunts in children: Recognition, evaluation, and management // Seminars Liver Dis. 2012. Vol. 32, N 4. P. 273–287. doi: 10.1055/s-0032-1329896
- Papamichail M., Pizanias M., Heaton N. Congenital portosystemic venous shunt // Eur J Pediatrics. 2018. Vol. 177, N 3. P. 285–294. doi: 10.1007/s00431-017-3058-x
- Abernethy J. Account of two instances of uncommon formation in the viscera of the human body: From the philosophical transactions of the royal society of London // Med Facts Observations. 1797. N 7. P. 100–108.
- Guérin F., Blanc T., Gauthier F., et al. Congenital portosystemic vascular malformations // Seminars Pediatric Sur. 2012. Vol. 21, N 3. P. 233–244. doi: 10.1053/j.sempedsurg.2012.05.006
- Born M. The ductus venosus // RoFo Fortschritte Gebiet Rontgenstrahlen Bildgebenden Verfahren. 2021. Vol. 193, N 5. P. 521–526. doi: 10.1055/a-1275-0984
- Baller S.E., Reinehr M., Haslinger C., et al. Case report of neonatal ductus venosus atresia // J Neonatal-Perinatal Med. 2021. Vol. 14, N 2. P. 307–312. doi: 10.3233/NPM-190398
- Franchi-Abella S., Branchereau S., Lambert V., et al. Complications of congenital portosystemic shunts in children: Therapeutic options and outcomes // J Pediatric Gastroenterol Nutrition. 2010. Vol. 51, N 3. P. 322–330. doi: 10.1097/MPG.0b013e3181d9cb92
- Morgan G., Superina R. Congenital absence of the portal vein: Two cases and a proposed classification system for portasystemic vascular anomalies // J Pediatric Sur. 1994. Vol. 29, N 9. P. 1239–1241. doi: 10.1016/0022-3468(94)90812-5
- Tang H., Song P., Wang Z., et al. A basic understanding of congenital extrahepatic portosystemic shunt: Incidence, mechanism, complications, diagnosis, and treatment // Intractable Rare Dis Res. 2020. Vol. 9, N 2. P. 64–70. doi: 10.5582/irdr.2020.03005
- Păcurar D., Dijmărescu I., Dijmărescu A.D., et al. A case report on an incidental discovery of congenital portosystemic shunt // Medicine. 2019. Vol. 98, N 31. P. e16679. doi: 10.1097/MD.0000000000016679
- Shah A., Aziz A., Awwad A., et al. Incidental radiological diagnosis of asymptomatic Abernethy malformations: Two case reports // BJR|Case Reports. 2017. Vol. 3, N 1. P. 20150496. doi: 10.1259/bjrcr.20220059
- Yangín-Ergon E., Ermis N., Colak R., et al. Abernethy malformation type 2 and biliary atresia coexistence: A rare cause of infantile liver transplant // Euroasian J Hepatogastroenterol. 2018. Vol. 8, N 2. P. 163–166. doi: 10.5005/jp-journals-10018-1283
- Sahu M.K., Bisoi A.K., Chander N.C., et al. Abernethy syndrome, a rare cause of hypoxemia: A case report // Ann Pediatric Cardiol. 2015. Vol. 8, N 1. P. 64–66. doi: 10.3389/fcvm.2021.784739
- Lux D., Naito A., Harikrishnan S. Congenital extrahepatic portosystemic shunt with progressive myelopathy and encephalopathy // Practical Neurology. 2019. Vol. 19, N 4. P. 368–371. doi: 10.1136/practneurol-2018-002111
- Merola E., Cao M., La Starza S., et al. Portosystemic encephalopathy in an 86-year-old patient: A clinical challenge // Acta Gastro-Enterologica Belgica. 2016. Vol. 79, N 1. P. 58–59.
- Sharma R., Suddle A., Quaglia A., et al. Congenital extrahepatic portosystemic shunt complicated by the development of hepatocellular carcinoma // Hepatobiliary Pancreatic Dis Int. 2015. Vol. 14, N 5. P. 552–557. doi: 10.1016/S1499-3872(15)60418-0
- Lin X., Rao J., Xiang Y., et al. Case report: A rare syncope case caused by abernethy ii and a review of the literature // Front Cardiovascul Med. 2022. Vol. 8. P. 2050. doi: 10.3389/fcvm.2021.784739
- Hasegawa T., Sato T., Ishii T., et al. Oral sodium phenylbutyrate for hyperammonemia associated with congenital portosystemic shunt: A case report // J Pediatric Endocrinology Metabolism. 2021. Vol. 34, N 3. P. 407–410. doi: 10.1515/jpem-2020-0603
- Peček J., Fister P., Homan M. Abernethy syndrome in Slovenian children: Five case reports and review of literature // World J Gastroenterol. 2020. Vol. 26, N 37. P. 5731–5744. doi: 10.3748/wjg.v26.i37.5731
- Pathak A., Agarwal N., Mandliya J., et al. Abernethy malformation: A case report // BMC Pediatrics. 2012. Vol. 12, N 1. P. 1. doi: 10.1186/1471-2431-12-57
- Duarte-Mesquita R., Sousa M., Vilaverde F., Cardoso R. Abernethy malformation: Beware in cases of unexplained hepatic encephalopathy in adults // BJR| Case Reports. 2017. Vol. 4, N 2. P. 20170054. doi: 10.1259/bjrcr.20170054
- Allegritti M., Enrico B., Basile E., et al. Non-cirrhotic extra-hepatic porto-systemic shunt causing adult-onset encephalopathy treated with endovascular closure // Digestive Diseases and Sciences. 2020. Vol. 65, N 4. P. 946–951. doi: 10.1007/s10620-019-06024-4
- Alvi A.A., Pichardo J., Gupta S., et al. An interesting case of congenital intrahepatic porto-hepatic shunt as a cause of unexplained encephalopathy // Cureus. 2020. Vol. 12, N 4. P. e7639. doi: 10.14309/01.ajg.0000598392.71372.f2
- De Vito C., Tyraskis A., Davenport M., et al. Histopathology of livers in patients with congenital portosystemic shunts (Abernethy malformation): A case series of 22 patients // Virchows Archiv. 2019. Vol. 474, N 1. P. 47–57. doi: 10.1007/s00428-018-2464-4
- Lautz T.B., Shah S.A., Superina R.A. Hepatoblastoma in children with congenital portosystemic shunts // J Pediatric Gastroenterology Nutrition. 2016. Vol. 62, N 4. P. 542–545. doi: 10.1097/MPG.0000000000001012
- Correa C., Luengas J.P., Howard S.C., Veintemilla G. Hepatoblastoma and abernethy malformation type I: Case report // J Pediatric Hematology/Oncology. 2017. Vol. 39, N 2. P. e79–e81. doi: 10.1097/MPH.0000000000000650
- Kwapisz L., Wells M.M., Judaibi B. Al Abernethy malformation: Congenital absence of the portal vein // Can J Gastroenterol Hepatol. 2014. Vol. 28, N 11. P. 587–588. doi: 10.1155/2014/675812
- Benedict M., Rodriguez-Davalos M., Emre S., et al. Congenital extrahepatic portosystemic shunt (abernethy malformation type Ib) with associated hepatocellular carcinoma: Case report and literature review // Pediatric Developmental Pathology. 2017. Vol. 20, N 4. P. 354–362. doi: 10.1177/1093526616686458
- Özden İ., Yavru A., Güllüoğlu M., et al. Transplantation for large liver tumors in the setting of abernethy malformation // Experimental Clin Transplantation. 2017. Vol. 15, Suppl. 2. P. 82–85. doi: 10.6002/ect.TOND16.L23
- Lin K. Y., Chen H., Yu L. Pulmonary arterial hypertension caused by congenital extrahepatic portocaval shunt: A case report // BMC Cardiovascular Disorders. 2019. Vol. 19, N 1. P. 1–5. doi: 10.1186/s12872-019-1124-1
- Osorio M.J., Bonow A., Bond G.J., et al. Abernethy malformation complicated by hepatopulmonary syndrome and a liver mass successfully treated by liver transplantation // Pediatric Transplantation. 2011. Vol. 15, N 7. P. 149–151. doi: 10.1111/j.1399-3046.2010.01337.x
- Baiges A., Turon F., Simón-Talero M., et al. Congenital extrahepatic portosystemic shunts (Abernethy malformation): An international observational study // Hepatology. 2020. Vol. 71, N 2. P. 658–669. doi: 10.1002/hep.30817
- Ponziani F.R., Faccia M., Zocco M.A., et al. Congenital extrahepatic portosystemic shunt: Description of four cases and review of the literature // J Ultrasound. 2019. N 22. P. 349–358. doi: 10.1007/s40477-018-0329-y
- Sheth R., Sivakumar K. The Abernethy malformation with inferior caval vein hypoplasia: A tailored technique for transcatheter closure and an insight into embryological perspective // Cardiology Young. 2018. Vol. 28, N 9. P. 1169–1171. doi: 10.1017/S1047951118000884
- Li H., Ma Z., Xie Y., Tian F. Recurrent hyperammonemia after abernethy malformation Type 2 closure: A case report // Ann Hepatol. 2017. Vol. 16, N 3. P. 460–464. doi: 10.5604/01.3001.0009.8603
- Kanazawa H., Nosaka S., Miyazaki O., et al. The classification based on intrahepatic portal system for congenital portosystemic shunts // J Pediatric Sur. 2015. Vol. 50, N 4. P. 688–695. doi: 10.1016/j.jpedsurg.2015.01.009
Дополнительные файлы

