







Magnetic resonance imaging for diagnosing a rare disease: incontinentia pigmenti (Bloch–Sulzberger syndrome) on the example of a clinical case
- Authors: Yarmola I.I.1, Anikin A.V.1, Gankin D.A.2, Fomina L.E.1, Kharitonova N.A.1, Zhanin I.S.1, Pushkov A.A.1, Basargina M.A.1, Kondakova O.B.1
-
Affiliations:
- National Medical Research Center for Children's Health
- Shchelkovsky Perinatal Center
- Issue: Vol 4, No 3 (2023)
- Pages: 384-392
- Section: Case reports
- Submitted: 16.05.2023
- Accepted: 29.06.2023
- Published: 26.09.2023
- URL: https://jdigitaldiagnostics.com/DD/article/view/430154
- DOI: https://doi.org/10.17816/DD430154
- ID: 430154

Cite item

Abstract
Incontinentia pigmenti, also known as Bloch–Sulzberger syndrome, is a rare hereditary disease characterized by typical skin rashes and involvement of other organs and systems. Magnetic resonance imaging stands as the primary method for visualizing the structural pathology of the brain and predicting neurological manifestations in an affected child.
Diagnosing incontinentia pigmenti predominantly falls within the domain of dermatologists; verification is performed by molecular genetic analysis of the IKBKG gene. This study involved magnetic resonance imaging of the brain in a patient with skin rashes, characteristic of Bloch–Sulzberger syndrome, and deletion in the IKBKG gene, where numerous foci of ischemia, hemorrhages, and lesions of the tracts were detected.
Magnetic resonance imaging of the brain in patients with Bloch–Sulzberger syndrome is used to evaluate the severity of damage to the brain substance, which makes it possible to explain the cause of neurological symptoms and correct habilitation, as well as predict the development of the child.
Full Text
BACKGROUND
Skin, hair, teeth, nails, eyes, and the central nervous system (CNS) are all damaged by incontinentia pigmenti (OMIM 308300: Familial male-lethal type Bloch–Sulzberger syndrome; incontinentia pigmenti type II). The condition typically appears in the first few days or weeks of life.
In the global literature, 2,000 cases of incontinentia pigmenti have been described, and the number is increasing. The global prevalence of incontinentia pigmenti is estimated to be 0.7 cases per million people. The incidence in Europe is 1.2 cases per 100,000 neonates [1]. Mutations in the inhibitor of kappa B kinase gamma (IKBKG) gene at position Xq28 on the Х chromosome’s long arm cause the disorder. The gene regulates apoptosis, cell cycle, inflammation, immunology, and other processes [2–4].
The condition has a wide spectrum of clinical manifestations, ranging from a mildly reduced quality of life to deadly results. The changes in the nucleotide sequence in the gene result in structural changes in the brain [5]. Magnetic resonance imaging (MRI) can detect anomalies, which is useful in identifying various monogenic [6] and multifactorial inherited disorders [7].
Incontinentia pigmenti is inherited in an X-linked dominant trait that kills most male embryos during development. The affected girl-to-boy ratio is 20:1.
Multiple organs and systems are involved in incontinentia pigmenti, most of which emerge from the ectoderm. In 100% of cases, several skin eruptions are found. The disease is distinguished by the linear alignment of the vesicles and pustules along the lines of Blaschko. Patients with inconintentia pigmenti have been documented to have alopecia, oral development anomalies, and fingernail dystrophy. Furthermore, affected patients are more likely to develop ocular pathologies, such as retinal hypervascularization, which can lead to retinal detachment if left untreated (typically occurs during the first 6 yr of life) [8]. Strabismus, cataracts, optic atrophy, retinal pigmentation, and microphthalmia have also been reported. According to several authors, the CNS is involved in 10% to 30% of cases [9], resulting in seizures of varying severity (cases ranging from a single seizure episode to chronic epilepsy), cognitive impairment, mental retardation, and spastic paresis. Less common disorders include breast aplasia, supernumerary nipples, primary pulmonary hypertension, and leukocytosis.
Molecular genetic testing identifies IKBKG mutations, and histological examination of the skin tissue samples is used to diagnose the condition. There is a clinical indication [10] to suspect pigmentary incontinence.
The treatment is symptomatic to prevent skin infections, retinal detachment, and epileptic seizures. Dental implants or orthodontic correction are recommended for dental problems, whereas therapy is recommended for spastic activity or paresis.
CASE REPORT
A 14-day-old girl was taken to the Neonatal and Early Age Pathology Department of the National Medical Research Center for the Children’s Health of the Ministry of Health of the Russian Federation.
Prenatal history
In the first and second trimesters of pregnancy, there was a threat of miscarriage due to cytomegalovirus infection (confirmed by polymerase chain reaction) in the first trimester; the mother had inpatient therapy. The mother experienced three episodes of acute respiratory virus infections in the third trimester and herpetic eruptions twice.
The infant was delivered at 38 weeks gestation as the second full-term spontaneous childbirth: body weight at birth, 3,470 g; body length, 53 cm; and Apgar score: 9/9.
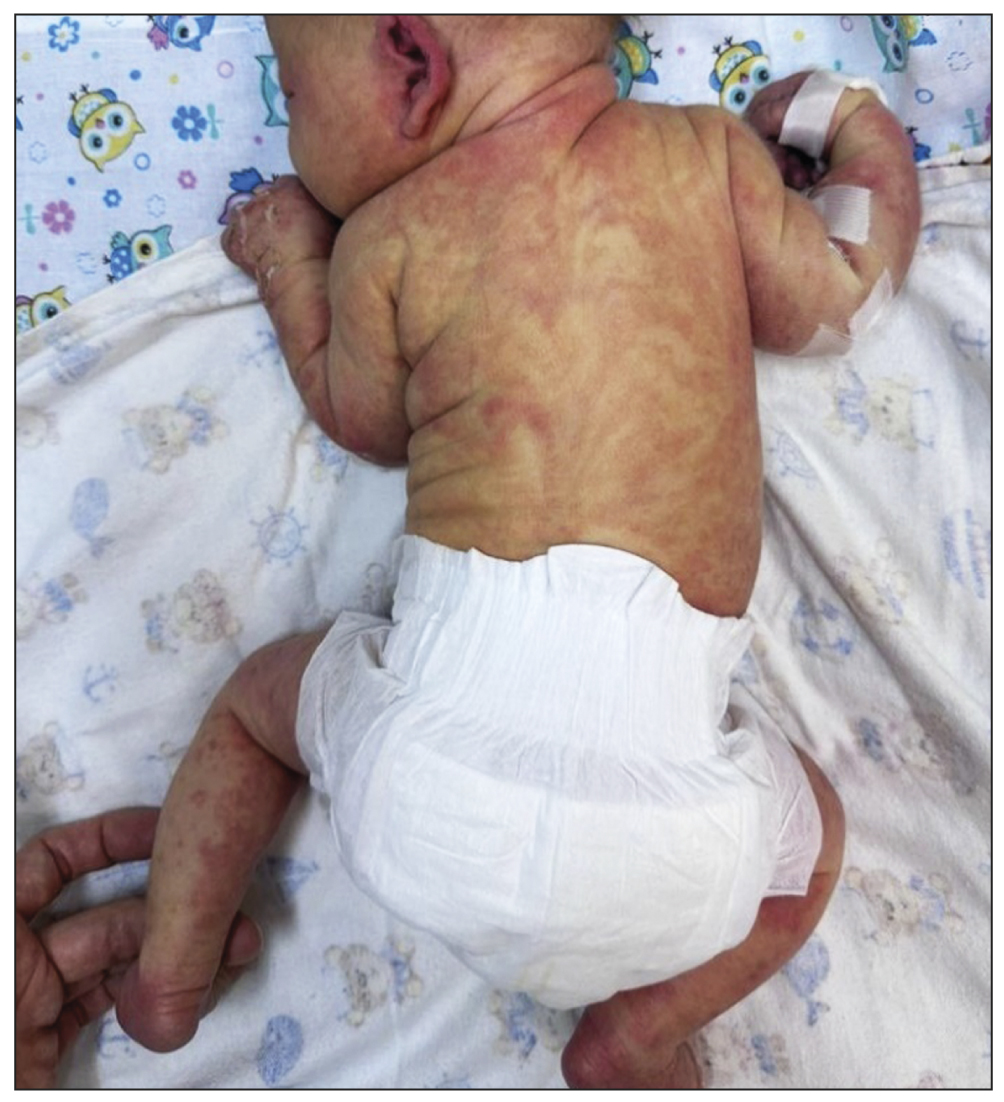
The post-natal condition was satisfactory. Extensive exanthema covered the entire body, including the trunk, face, and limbs (Figure 1). The eruption had been removed from the scalp. The laboratory results were age-appropriate; the inflammatory indicators were not present.

Fig.1. Vesicles aligned with the lines of Blaschko.
The general condition began to deteriorate on Day 4: cyanotic spots appeared on the limbs; the skin was icteric, with a grayish hue; the breathing became weakened, with a tendency to tachypnea; low blood oxygen saturation (SpO2: 81%–95%) was determined; hyperexcitability at examinations and depressed consciousness at rest were observed; upper and lower limb muscles were hypertonic; and head lag as well as seizures were demonstrated.
Laboratory data showed the following changes: blood pH reduced to 7.242, blood lactate increased to 6.4 mmol/L, and HCO3 decreased to 16.7 mmol/L (metabolic acidosis).
Dermatological examination revealed that the skin lesions had receded and that linear brown-to-light pink pigmentation had appeared, which had turned to hypopigmentation during the follow-up. Bloch–Sulzberger syndrome was the preliminary diagnosis.
Molecular genetic testing was run to identify exons 4–10 deletion in IKBKG: multiplex allele-specific polymerase chain reaction with primers as described by Haque et al. [11] was used. The findings revealed heterozygous deletion of IKBKG exons 4–10 in patients with the Bloch–Sulzberger syndrome, as published in the Human Gene Mutation Database Professional. This frequent mutation in incontinentia pigmenti occurs in 65% of patients [12].
On Day 4, the girl’s condition was assessed as moderately severe due to the syndrome of CNS depression. Neurosonography revealed extensive ischemia abnormalities in the brain parenchyma and numerous focal changes in the parasagittal and periventricular areas.
On Day 7, an MRI indicated large areas of tiny focal lesions in the cerebral hemispheres, with secondary involvement of the corpus callosum and the corticospinal tract. The findings were interpreted as the result of repeated ischemic strokes induced by incontinentia pigmenti (Bloch–Sulzberger syndrome).
DISCUSSION
Bloch–Sulzberger syndrome is caused by a genetic abnormality that increases the sensitivity of cells developing from ectoderm to cytokine effects, resulting in apoptosis [13]. The skin and its derivatives (nails, hair, and teeth) and the nervous system are formed from the ectoderm. Disease symptoms commonly arise within the first 10 days of life. Because the disease stage at birth varies, the previous stages are assumed to occur in utero.
The disease manifested in the described patient in a typical manner during the first days of life, beginning with linear skin eruptions, which later experienced incremental modifications in accordance with the disease pathomorphosis and were accompanied by neurological symptoms.
Multiple small (min. 2 mm) restricted diffusion lesions were found in the deep white matter, in and under the cortex, in the corpus callosum, posterior limb of the internal capsule, cerebral peduncles, along the corticospinal tract, and other tracts (Figure 2). Changes in the spinal pathways can be interpreted as either direct injury from incontinentia pigmenti or an early indicator of Wallerian degeneration (pre-Wallerian condition). The latter is characterized by spinal tract injury caused by neuron death and the degradation of the myelin sheath [14]. Small patches of restricted diffusion were regarded as tissue necrosis (infarctions).

Fig. 2. Diffuse-weighted brain images in the axial plane: (a) the arrow shows the hyperintense signal from the spinal tracts in the brain peduncles and (b) multiple lesions and involvement of the corpus callosum.
There are reports in the literature of minor focal brain infarctions caused by injury to the walls of small and medium-sized arteries, resulting in microhemorrhages and thrombosis. There have also been reports of extensive bilateral hemorrhagic necrosis cases with broad brain tissue damage [15].
Among the numerous lesions in the presented instance, the brain MRI revealed single regions with hemorrhagic features compatible with ischemia (Figure 3а). This suggests that not all ischemia foci were accompanied by bleeding. Although necrotic areas eventually turn into encephalomalacia areas, some areas with minimal damage may recover entirely and even be consistent with the normal structure of brain matter in MRI [16]. The hyperintense signal was seen in the cortical area and at the gray/white matter interface in the frontal and parietal lobes in T1-weighted images (T1-WI) (Figure 3b). These areas resemble cortical necrosis, which occurs after ischemia damage to the cortex, resulting in monocyte infiltration and phagocytosis of cell fragments in the damaged structures. T1 images are hyperintense due to deposits of denatured protein of the dead cells and/or lipid-loaded macrophages [17].

Fig. 3. Magnetic resonance imaging (MRI) of the brain: (a) susceptibility-weighted images (the arrows show microhemorrhages) and (b) Т1-weighted images (the arrows indicate hyperintense areas of the cortical necrosis).
Furthermore, histologically validated cases of inflammatory changes of the arachnoid mater and the brain with eosinophilic infiltration in patients with incontinentia pigmenti have been documented, mimicking infection-related damage without evident vascular disorders [18].
First, an MRI-based differential diagnosis is made for encephalitis and neonatal hypoxic–ischemic damage. Encephalitis is distinguished by alternating short and long areas of hyperintense signal in T2-WI with possible restricted or hyperintense diffusion, whereas incontinentia pigmenti is distinguished by chaotic small-lesion damage primarily localized in the white matter. The perinatal ischemia image corresponds to brain structural development patterns; full-term neonates generally have periventricular leukomalacia, basal ganglia damage, or violations that follow the artery patterns. Differential diagnosis of embolic stroke is complex and requires a comprehensive approach, with medical history collection and patient examination.
Vesicular rash and neurological signs may be mistaken for manifestations of herpes infection. In this case, MRI results, typical skin eruptions with evident stages of pathomorphosis, and a negative test for herpes infection should prompt clinicians to consider incontinentia pigmenti.
CONCLUSION
Because the skin eruptions are specific and follow clear stages, a dermatologist is essential in identifying incontinentia pigmenti. The molecular genetic test for IKBKG mutations is also important in diagnosis.
Brain MRI is the first imaging tool to assess brain matter injury when patients with Bloch–Sulzberger syndrome develop neurological symptoms. This method is safe for dynamic follow-up and enables objectively assessing rehabilitation potential, correcting the rehabilitation plan, and predicting the child’s development.
ADDITIONAL INFORMATION
Funding source. This article was not supported by any external sources of funding.
Competing interests. The authors declare that they have no competing interests.
Authors’ contribution. All authors made a substantial contribution to the conception of the work, acquisition, analysis, interpretation of data for the work, drafting and revising the work, final approval of the version to be published and agree to be accountable for all aspects of the work. I.I. Yarmola ― writing text of the article, planning article structure, imaging analysis; A.V. Anikin ― editing of the article, analytical work, discussion of the results, read and approved the direction of the manuscript for publication; D.A. Gankin ― editing the text of the article, discussion of the results; L.E. Fomina ― edition of the text of the article; N.A. Kharitonova ― read and approved the direction of the manuscript for publication; I.S. Zhanin, A.A. Pushkov ― article editing, conducting molecular genetic analysis; O.B. Kondakova ― article editing, genetic counseling of the patient.
Consent for publication. Written consent was obtained from the patient’s parents for publication of relevant medical information and all of accompanying images within the manuscript in Digital Diagnostics Journal.
About the authors
Igor I. Yarmola
National Medical Research Center for Children's Health
Author for correspondence.
Email: lord_dukich@bk.ru
ORCID iD: 0000-0002-1272-5119
SPIN-code: 5591-8066
Russian Federation, Moscow
Anatoly V. Anikin
National Medical Research Center for Children's Health
Email: anikacor@gmail.com
ORCID iD: 0000-0003-0362-6511
SPIN-code: 7592-1352
MD, Cand. Sci. (Med.)
Russian Federation, MoscowDmitry A. Gankin
Shchelkovsky Perinatal Center
Email: ganja-nn@yandex.ru
ORCID iD: 0009-0001-6779-8702
Russian Federation, Schelkovo
Lyubov E. Fomina
National Medical Research Center for Children's Health
Email: love.fomina@mail.ru
ORCID iD: 0000-0002-3838-3284
SPIN-code: 1298-8350
Russian Federation, Moscow
Natalia A. Kharitonova
National Medical Research Center for Children's Health
Email: kharitonovan@nczd.ru
ORCID iD: 0000-0002-6912-1471
SPIN-code: 7379-8269
MD, Cand. Sci. (Med.)
Russian Federation, MoscowIlya S. Zhanin
National Medical Research Center for Children's Health
Email: zhaninis@nczd.ru
ORCID iD: 0000-0003-1423-0379
SPIN-code: 6108-2016
MD, Cand. Sci. (Med.)
Russian Federation, MoscowAleksandr A. Pushkov
National Medical Research Center for Children's Health
Email: n1972z@yandex.ru
ORCID iD: 0000-0001-6648-2063
SPIN-code: 2928-5764
Cand. Sci. (Biol.)
Russian Federation, MoscowMilana A. Basargina
National Medical Research Center for Children's Health
Email: kharitonovan@nczd.ru
ORCID iD: 0000-0003-2075-6668
SPIN-code: 5504-7154
MD, Cand. Sci. (Med.)
Russian Federation, MoscowOlga B. Kondakova
National Medical Research Center for Children's Health
Email: n1972z@yandex.ru
ORCID iD: 0000-0002-6316-9992
SPIN-code: 9066-3698
MD, Cand. Sci. (Med.)
Russian Federation, MoscowReferences
- Scheuerle AE, Ursini MV, Adam MP, et al. Incontinentia Pigmenti. In: GeneReviews [Internet], Seattle (WA): University of Washington, Seattle; 1993.
- Fusco F. Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-B activation. Hum Mol Genet. 2004;13(16):1763–1773. doi: 10.1093/hmg/ddh192
- Yadlapati S, Tripathy K. Incontinentia pigmenti (Bloch Sulzberger Syndrome). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023.
- Savostyanov KV. Modern algorithms of genetic diagnosis of rare hereditary diseases in Russian patients. Moscow: Polygraphist Publisher; 2022. 451 р. (In Russ).
- Minić S, Cerovac N, Novaković I, et al. The impact of the IKBKG gene on the appearance of the corpus callosum abnormalities in incontinentia pigmenti. Diagnostics. 2023;13(7):1300. doi: 10.3390/diagnostics13071300
- Chistiakov DA, Savostanov KV, Kuzenkova LM, et al. Molecular characteristics of patients with glycosaminoglycan storage disorders in Russia. Clin Chim Acta. 2014;(436):112–120. doi: 10.1016/j.cca.2014.05.010
- Chistyakov DA, Savostanov KV, Nosikov VV, Turakulov RI. Genetic determinants of Graves’ disease. Mol Gen Metabol. 2000;71(1-2):66–69. doi: 10.1006/mgme.2000.3042
- Meuwissen ME, Mancini GM. Neurological findings in incontinentia pigmenti: A review. Eur J Med Genet. 2012;55(5):323–331. doi: 10.1016/j.ejmg.2012.04.007
- Carney RG. Incontinentia pigmenti. A world statistical analysis. Arch Dermatol. 1976;112(4):535–542.
- Minić S, Trpinac D, Obradović M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85(6):536–542. doi: 10.1111/cge.12223
- Haque MN, Ohtsubo M, Nishina S, et al. Analysis of IKBKG/NEMO gene in five Japanese cases of incontinentia pigmenti with retinopathy: Fine genomic assay of a rare male case with mosaicism. J Hum Genet. 2021;66(2):645–645. doi: 10.1038/s10038-020-00836-3
- Kawai M, Kato T, Tsutsumi M, et al. Molecular analysis of low-level mosaicism of the IKBKG mutation using the X Chromosome Inactivation pattern in Incontinentia Pigmenti. Mol Genet Genomic Med. 2020;8(12):e1531. doi: 10.1002/mgg3.1531
- Tak PP, Firestein GS. NF-κB: A key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11. doi: 10.1172/JCI11830
- Kleinman JT. Early Wallerian degeneration on magnetic resonance imaging: Underappreciated but highly relevant. Dev Med Child Neurol. 2013;55(2):104–105. doi: 10.1111/dmcn.12022
- Salamon SA, Lichtenbelt K, Cowan FM, et al. Clinical presentation and spectrum of neuroimaging findings in newborn infants with incontinentia pigmenti. Dev Med Child Neurol. 2016;58(10):1076–1084. doi: 10.1111/dmcn.13140
- Lou H, Zhang L, Xiao W, et al. Nearly completely reversible brain abnormalities in a patient with incontinentia pigmenti. Am J Neuroradiol. 2008;29(3):431–433. doi: 10.3174/ajnr.A0890
- Kinoshita T, Ogawa T, Yoshida Y, et al, Curvilinear T1 hyperintense lesions representing cortical necrosis after cerebral infarction. Neuroradiology. 2005;47(7):647–651. doi: 10.1007/s00234-005-1398-0
- Hauw JJ, Perié G, Bonnette J, Escourolle R. [Neuropathological study of incontinentia pigmenti. Anatomical case report (author’s transl). (In French)]. Acta Neuropathol. 1977;38(2):159–162. doi: 10.1007/BF00688564
Supplementary files

