

")








Магнитно-резонансная томография в диагностике редкого генетического заболевания ― недержания пигмента (синдром Блоха–Сульцбергера) ― на примере клинического случая
- Авторы: Ярмола И.И.1, Аникин А.В.1, Ганькин Д.А.2, Фомина Л.Е.1, Харитонова Н.А.1, Жанин И.С.1, Пушков А.А.1, Басаргина М.А.1, Кондакова О.Б.1
-
Учреждения:
- Национальный медицинский исследовательский центр здоровья детей
- Щёлковский перинатальный центр
- Выпуск: Том 4, № 3 (2023)
- Страницы: 384-392
- Раздел: Клинические случаи и серии клинических случаев
- Статья получена: 16.05.2023
- Статья одобрена: 29.06.2023
- Статья опубликована: 26.09.2023
- URL: https://jdigitaldiagnostics.com/DD/article/view/430154
- DOI: https://doi.org/10.17816/DD430154
- ID: 430154

Цитировать

Аннотация
Недержание пигмента (синдром Блоха–Сульцбергера) ― редкое наследственное заболевание, проявляющееся характерными кожными высыпаниями и поражением других органов и систем. Магнитно-резонансная томография является приоритетным методом для визуализации структурной патологии головного мозга и прогноза неврологической манифестации у ребёнка.
Ключевая роль диагностики заболевания недержания пигмента принадлежит дерматологу; подтверждающая диагностика проводится путём молекулярно-генетического анализа гена IKBKG.
В представленном клиническом наблюдении у новорождённой девочки с высыпаниями на кожных покровах, типичными для синдрома Блоха–Сульцбергера, и выявленной делецией в гене IKBKG проводилась магнитно-резонансная томография головного мозга, где были обнаружены многочисленные очаги ишемии, кровоизлияния и поражение проводящих путей.
Магнитно-резонансная томография головного мозга у пациентов с синдромом Блоха–Сульцбергера используется для оценки тяжести поражения вещества мозга, что позволяет объяснить причину неврологических симптомов, скорректировать реабилитационные мероприятия, а также прогнозировать развитие ребёнка.
Полный текст
АКТУАЛЬНОСТЬ
Недержание пигмента (OMIM 308300: синдром Блоха–Сульцбергера, недержание пигмента, тип II) ― редкое наследственное Х-сцепленное заболевание из группы генодерматозов, которое характеризуется поражением кожи, волос, зубов, ногтей, глаз и центральной нервной системы. Заболевание в большинстве случаев проявляется в первые дни или недели жизни.
В мировой литературе описано более 2000 случаев недержания пигмента, и их количество продолжает расти. Распространённость недержания пигмента оценивается в 0,7 случаев на 1 млн человек во всём мире. Частота заболевания на территории Европы ― 1,2:100 тыс. новорождённых [1]. Причиной заболевания являются патогенные варианты в гене IKBKG (ингибитор каппа-В киназы гамма), расположенном на длинном плече Х-хромосомы в локусе Xq28. Ген участвует в регуляции апоптоза, клеточного цикла, воспаления, иммунитета и других путей [2–4].
Для заболевания характерен широкий клинический полиморфизм ― от небольшого снижения качества жизни до летальных случаев. Патогенные нуклеотидные варианты в этом гене приводят к структурным изменениям головного мозга [5]. Эти аномалии могут быть выявлены при помощи магнитно-резонансной томографии (МРТ), используемой для диагностики и других наследственных болезней как моногенной [6], так и мультифакториальной природы [7].
Заболевание наследуется по Х-сцепленному доминантному типу и является летальным для большинства мальчиков в период эмбриогенеза. Соотношение больных девочек и мальчиков ― 20:1.
При недержании пигмента отмечается множественное поражение органов и систем, развивающихся преимущественно из эктодермальной пластинки. Высыпания на коже встречаются в 100% случаев и носят множественный характер. Отличительной особенностью является линейное расположение (по линиям Блашко) пузырей и пустул. У пациентов с недержанием пигмента описаны алопеция, аномалии развития зубов, дистрофия ногтей, а также повышение риска офтальмологической патологии ― гиперваскуляризации сетчатки, которая без лечения может приводить к отслоению (как правило, у таких пациентов отслоение происходит в возрасте до 6 лет жизни) [8]. Встречаются также косоглазие, катаракта, атрофия зрительных нервов, пигментная патология сетчатки и микрофтальм. Центральная нервная система, по данным разных авторов, поражается в 10–30% случаев [9] и проявляется судорогами различной степени тяжести (описаны случаи от единичного судорожного приступа до хронической эпилепсии), снижением когнитивных способностей, задержкой развития, спастическими парезами. Реже встречаются аплазия молочных желёз, дополнительные соски, первичная лёгочная гипертензия, лейкоцитоз и другие, более редкие патологические состояния.
Заболевание диагностируют молекулярно-генетическими методами, которые выявляют патогенные генетические варианты в гене IKBKG, а также с помощью гистологического исследования биоптатов кожи. Существуют и клинические критерии [10], которые позволяют заподозрить недержание пигмента.
Лечение носит симптоматический, превентивный характер и направлено на предотвращение кожных инфекций, отслойки сетчатки, купирование эпилептических приступов. Показаны имплантация и коррекция форм при поражении зубов, реабилитация при наличии спастических проявлений или парезов.
ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ
Ребёнок Л. (девочка) в возрасте 14 дней поступила в отделение патологии новорождённых и детей раннего детского возраста ФГАУ «НМИЦ здоровья детей» Минздрава России.
Пренатальный анамнез. Беременность протекала с угрозой прерывания в I и II триместрах вследствие инфицирования цитомегаловирусом (подтверждено методом полимеразной цепной реакции) в I триместре; проводилось стационарное лечение матери. В III триместре мама трижды болела острыми респираторными вирусными инфекциями, дважды появлялись герпетические высыпания.
Роды вторые, срочные, самопроизвольные на сроке гестации 38 недель; масса тела при рождении 3470 г, рост 53 см, оценка по шкале Апгар 9/9 баллов.
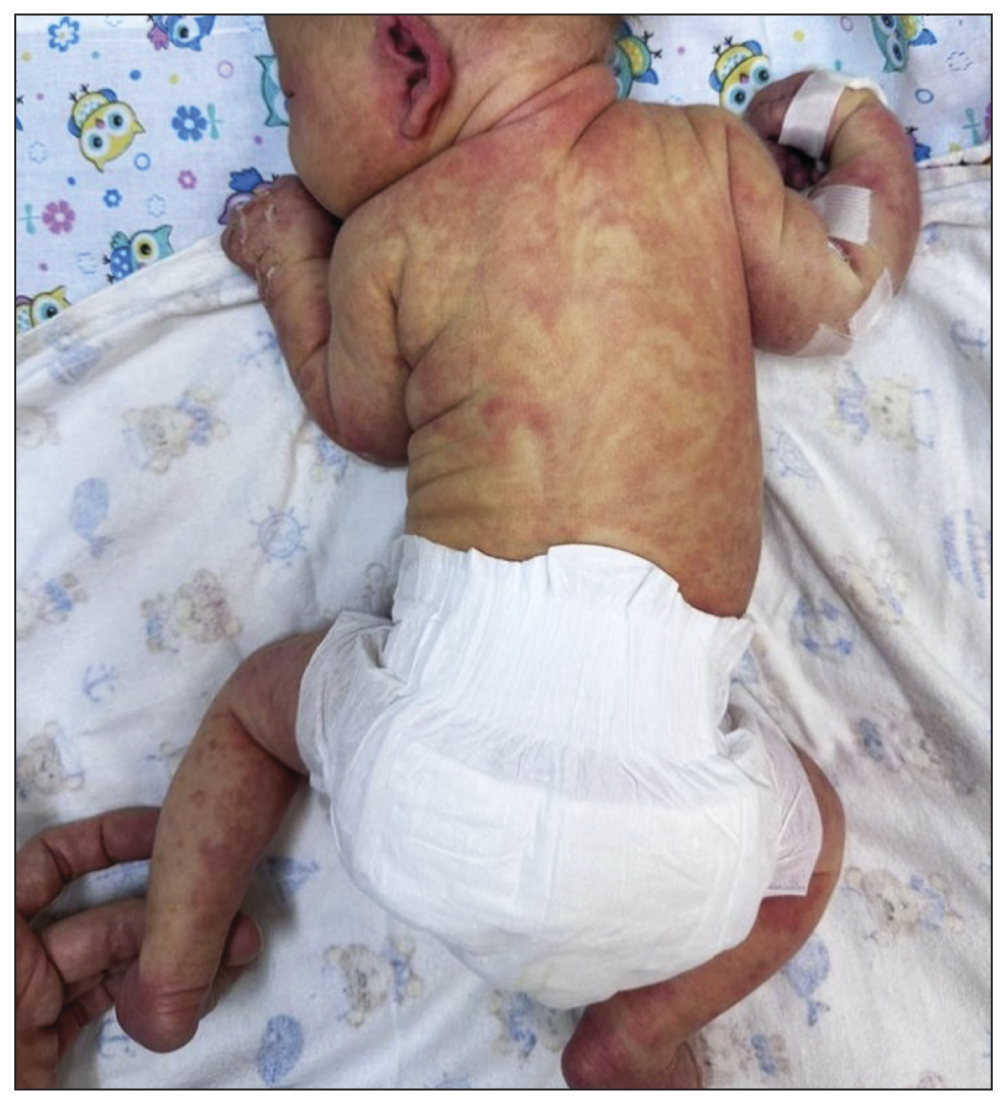
После рождения общее состояние удовлетворительное. Определялась распространённая экзантема по всему туловищу, лицу и конечностям (рис. 1). Область волосистой части головы свободна от высыпаний. Лабораторные показатели в пределах возрастной нормы, маркеры воспаления отсутствовали.

Рис. 1. Везикулы, распространяющиеся по линиям Блашко.
С четвёртых суток жизни отмечалось ухудшение общего состояния: появление цианотичных пятен на ногах и руках, иктеричности кожных покровов с сероватым оттенком, ослабление дыхания со склонностью к тахи-пное, низкое насыщение крови кислородом (сатурация, SpO2) ― 81–95%, развитие гипервозбудимости при осмотре и угнетения сознания в покое, гипертонуса мышц верхних и нижних конечностей, запрокидывания головы назад, судорог.
Отмечены изменения в анализах крови: снижение кислотности крови (рН) до 7,242, повышение лактата крови до 6,4 ммоль/л, снижение концентрации гидрокарбоната (НСО3-) до 16,7 ммоль/л (метаболический ацидоз).
При осмотре дерматологом отмечена динамика в виде регрессии очагов на коже с формированием пигментации коричневатого и светло-розового цвета линейной формы, с переходом в гипопигментацию при дальнейшем наблюдении. Выставлен предварительный диагноз: «Синдром Блоха–Сульцбергера».
Молекулярно-генетическое исследование было направлено на поиск делеции экзонов 4–10 в гене IKBKG методом мультиплексной аллельспецифичной полимеразной цепной реакции с использованием праймеров, описанных в статье M.N. Haque и соавт. [11]. В результате исследования обнаружена гетерозиготная делеция экзонов 4–10 в гене IKBKG, описанная в базе данных мутаций генов человека Human Gene Mutation Database (HGMD) Professional у пациентов с синдромом Блоха–Сульцбергера. Данный нуклеотидный вариант является одним из самых распространённых при недержании пигмента и встречается у 65% пациентов [12].
На четвёртые сутки жизни состояние ребёнка оценивалось как среднетяжёлое в связи с синдромом угнетения центральной нервной системы. По результатам нейросонографии, эхо-признаки диффузных ишемических изменений паренхимы, парасагиттальные и перивентрикулярные множественные очаговые изменения.
Магнитно-резонансная томография (МРТ) проведена на седьмые сутки: выявлены распространённые участки мелкоочагового поражения вещества больших полушарий с вторичным вовлечением проводящих путей ― мозолистого тела и кортикоспинальных трактов. Изменения расценены как последствия множественных инфарктов головного мозга в рамках заболевания недержания пигмента (синдром Блоха–Сульцбергера).
ОБСУЖДЕНИЕ
Генетический дефект при синдроме Блоха–Сульцбергера приводит к повышенной уязвимости клеток ― эмбриональных производных эктодермальной пластинки ― в виде апоптоза при воздействии цитокинов [13]. К таким тканям относятся кожные покровы с их дериватами (ногти, волосы, зубы), а также нервная система. Манифестация заболевания обычно происходит в первые десять суток жизни, однако младенец может родиться с любой стадией: в этом случае предполагается, что предыдущие стадии произошли внутриутробно.
У пациента Л. манифестация заболевания произошла в первые сутки жизни, типично, с появления кожных высыпаний линейного характера, которые с течением времени претерпевали характерный патоморфоз, соответствующий стадиям заболевания, в сочетании с неврологической симптоматикой.
При МРТ головного мозга были выявлены множественные мелкие (минимальный размер 2 мм) очаги с ограничением диффузии, которые располагались хаотично в глубоком белом веществе, в коре и субкортикально, в мозолистом теле, заднем бедре внутренней капсулы, в ножках мозга, а также по ходу кортикоспинальных и других трактов (рис. 2). Изменения проводящих путей могут рассматриваться как прямое поражение при недержании пигмента либо как проявление ранней Валлеровской (пре-Валлеровской) дегенерации. Последняя представляет собой повреждение проводящих трактов из-за гибели аксона и распада миелиновой оболочки [14]. Мелкие участки с ограничением диффузии расценивались нами как некроз ткани (инфаркты).

Рис. 2. Диффузионно-взвешенные изображения головного мозга в аксиальной плоскости: a ― стрелкой указано повышение сигнала от проводящих путей в ножках мозга; b ― множественные очаги и поражение мозолистого тела.
В литературе описаны механизмы мелкоочаговых инфарктов мозга, связанных с повреждением стенки сосудов среднего и мелкого калибра, что приводило к микрокровоизлияниям, тромбозам; описаны также случаи массивного двустороннего геморрагического некроза с генерализованным разрушением тканей головного мозга [15].
В представленном нами случае на МРТ головного мозга среди множества очагов были выявлены лишь несколько участков с элементами геморрагической трансформации, соответствующие зонам ишемии (рис. 3, а). Это означает, что не все ишемические очаги осложнились кровоизлияниями. Участки некроза в дальнейшем трансформируются в зоны энцефаломаляции, однако часть минимально поражённых участков может восстановиться практически полностью вплоть до нормальной структуры вещества мозга на МРТ [16]. В области коры и на границе белого и серого вещества в лобных и теменных долях отмечались участки повышенного сигнала на Т1-взвешенных изображениях (см. рис. 3, b). Такие участки имеют сходство с кортикальным некрозом, который возникает при ишемическом повреждении коры и приводит к моноцитарной инфильтрации и фагоцитозу клеточных фрагментов, повреждённых структур. Повышение сигнала на Т1 происходит из-за отложения денатурированного белка умерших клеток и/или макрофагов, насыщенных липидами [17].

Рис. 3. Магнитно-резонансная томография головного мозга: a ― SWI-изображения (взвешенные по магнитной восприимчивости) (стрелками указаны участки микрокровоизлияний); b ― Т1-взвешенные изображения (стрелками указаны гиперинтенсивные участки кортикального некроза).
Кроме того, в литературе описаны гистологически подтверждённые случаи воспалительных изменений мягкой паутинной оболочки и головного мозга с наличием клеточной (эозинофильной) инфильтрации у пациентов с недержанием пигмента, напоминающие инфекционное поражение без явных сосудистых нарушений [18].
Дифференциальная диагностика по данным МРТ проводится прежде всего с энцефалитом и перинатальным ишемически-гипоксическим поражением. Для энцефалита характерно преимущественное поражение коры в виде малопротяжённых-протяжённых зон повышения МР-сигнала на Т2-ВИ с возможным ограничением или усилением диффузии, в то время как при недержании пигмента происходит хаотичное мелкоочаговое поражение, локализованное в большей степени в белом веществе. Перинатальная ишемия проявляется соответственно паттернам структурной зрелости мозга, для доношенных детей характерны поражение в виде перивентрикулярной лейкомаляции, поражение базальных ганглиев либо инфаркты, соответствующие артериальным бассейнам. Дифференциальная диагностика с эмболическими ин-фарктами затруднительна и требует комплексного подхода с изучением анамнеза и осмотром пациента.
Везикулярная стадия сыпи и манифестация неврологических проявлений может быть ошибочно принята за проявления герпетической инфекции. В таком случае совокупность находок на МРТ, характерные кожные высыпания со стадийным патоморфозом и отрицательный анализ на герпесвирусную инфекцию с высокой вероятностью позволяют заподозрить недержание пигмента.
ЗАКЛЮЧЕНИЕ
Ключевая роль диагностики заболевания недержания пигмента принадлежит дерматологу из-за специфического характера высыпаний на коже и их стадийности. Важную роль в подтверждающей диагностике играет молекулярно-генетический анализ гена IKBKG.
МРТ головного мозга у пациентов с синдромом Блоха–Сульцбергера является приоритетным методом для оценки тяжести поражения вещества мозга при возникновении неврологической симптоматики. Метод является безопасным для динамического наблюдения, позволяет объективно оценить реабилитационный потенциал, скорректировать абилитационные мероприятия, а также спрогнозировать развитие ребёнка.
ДОПОЛНИТЕЛЬНО
Источник финансирования. Авторы заявляют об отсутствии внешнего финансирования при проведении работы.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Вклад авторов. Все авторы подтверждают соответствие своего авторства международным критериям ICMJE (все авторы внесли существенный вклад в разработку концепции, проведение работы и подготовку статьи, прочли и одобрили финальную версию перед публикацией). Наибольший вклад распределён следующим образом: И.И. Ярмола ― написание текста рукописи, составление плана статьи, анализ диагностических изображений; А.В. Аникин ― редактирование статьи, аналитическая работа, обсуждение результатов, одобрение и направление рукописи на рассмотрение; Д.А. Ганькин ― редактирование текста статьи, обсуждение результатов; Л.Е. Фомина ― редактирование текста статьи; Н.А. Харитонова, М.А. Басаргина ― одобрение и направление рукописи на рассмотрение к публикации; И.С. Жанин, А.А. Пушков ― редактирование текста статьи, проведение молекулярно-генетического анализа; О.Б. Кондакова ― редактирование текста статьи, генетическая консультация пациента.
Информированное согласие на публикацию. Авторы получили письменное согласие законных представителей пациента на публикацию медицинских данных и фотографий в журнале Digital Diagnostics.
ADDITIONAL INFORMATION
Funding source. This article was not supported by any external sources of funding.
Competing interests. The authors declare that they have no competing interests.
Authors’ contribution. All authors made a substantial contribution to the conception of the work, acquisition, analysis, interpretation of data for the work, drafting and revising the work, final approval of the version to be published and agree to be accountable for all aspects of the work. I.I. Yarmola ― writing text of the article, planning article structure, imaging analysis; A.V. Anikin ― editing of the article, analytical work, discussion of the results, read and approved the direction of the manuscript for publication; D.A. Gankin ― editing the text of the article, discussion of the results; L.E. Fomina ― edition of the text of the article; N.A. Kharitonova ― read and approved the direction of the manuscript for publication; I.S. Zhanin, A.A. Pushkov ― article editing, conducting molecular genetic analysis; O.B. Kondakova ― article editing, genetic counseling of the patient.
Consent for publication. Written consent was obtained from the patient’s parents for publication of relevant medical information and all of accompanying images within the manuscript in Digital Diagnostics journal.
Об авторах
Игорь Игоревич Ярмола
Национальный медицинский исследовательский центр здоровья детей
Автор, ответственный за переписку.
Email: lord_dukich@bk.ru
ORCID iD: 0000-0002-1272-5119
SPIN-код: 5591-8066
Россия, Москва
Анатолий Владимирович Аникин
Национальный медицинский исследовательский центр здоровья детей
Email: anikacor@gmail.com
ORCID iD: 0000-0003-0362-6511
SPIN-код: 7592-1352
канд. мед. наук
Россия, МоскваДмитрий Александрович Ганькин
Щёлковский перинатальный центр
Email: ganja-nn@yandex.ru
ORCID iD: 0009-0001-6779-8702
Россия, Щёлково
Любовь Евгеньевна Фомина
Национальный медицинский исследовательский центр здоровья детей
Email: love.fomina@mail.ru
ORCID iD: 0000-0002-3838-3284
SPIN-код: 1298-8350
Россия, Москва
Наталия Александровна Харитонова
Национальный медицинский исследовательский центр здоровья детей
Email: kharitonovan@nczd.ru
ORCID iD: 0000-0002-6912-1471
SPIN-код: 7379-8269
канд. мед. наук
Россия, МоскваИлья Сергеевич Жанин
Национальный медицинский исследовательский центр здоровья детей
Email: zhaninis@nczd.ru
ORCID iD: 0000-0003-1423-0379
SPIN-код: 6108-2016
канд. мед. наук
Россия, МоскваАлександр . Алексеевич Пушков
Национальный медицинский исследовательский центр здоровья детей
Email: n1972z@yandex.ru
ORCID iD: 0000-0001-6648-2063
SPIN-код: 2928-5764
канд. биол. наук
Россия, МоскваМилана Александровна Басаргина
Национальный медицинский исследовательский центр здоровья детей
Email: kharitonovan@nczd.ru
ORCID iD: 0000-0003-2075-6668
SPIN-код: 5504-7154
канд. мед. наук
Россия, МоскваОльга Борисовна Кондакова
Национальный медицинский исследовательский центр здоровья детей
Email: n1972z@yandex.ru
ORCID iD: 0000-0002-6316-9992
SPIN-код: 9066-3698
канд. мед. наук
Россия, МоскваСписок литературы
- Scheuerle A.E., Ursini M.V., Adam M.P., et al. Incontinentia pigmenti. In: GeneReviews [Internet], Seattle (WA): University of Washington, Seattle, 1993.
- Fusco F. Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-B activation // Hum Mol Genet. 2004. Vol. 13, N 16. Р. 1763–1773. doi: 10.1093/hmg/ddh192
- Yadlapati S., Tripathy K. Incontinentia pigmenti (Bloch Sulzberger Syndrome). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing, 2023.
- Савостьянов К.В. Современные алгоритмы генетической диагностики редких наследственных болезней у российских пациентов. Москва: Полиграфист и издатель, 2022. 451 с.
- Minić S., Cerovac N., Novaković I., et al. The impact of the IKBKG gene on the appearance of the corpus callosum abnormalities in incontinentia pigmenti // Diagnostics. 2023. Vol. 13, N 7. Р. 1300. doi: 10.3390/diagnostics13071300
- Chistiakov D.A., Savostanov K.V., Kuzenkova L.M., et al. Molecular characteristics of patients with glycosaminoglycan storage disorders in Russia // Clin Chim Acta. 2014. N 436. Р. 112–120. doi: 10.1016/j.cca.2014.05.010
- Chistyakov D.A., Savostanov K.V., Nosikov V.V., Turakulov R.I. Genetic determinants of Graves’ disease // Mol Gen Metabol. 2000. Vol. 71, N 1-2. P. 66–69. doi: 10.1006/mgme.2000.3042
- Meuwissen M.E., Mancini G.M. Neurological findings in incontinentia pigmenti: A review // Eur J Med Genet. 2012. Vol. 55, N 5. Р. 323–331. doi: 10.1016/j.ejmg.2012.04.007
- Carney R.G. Incontinentia pigmenti. A world statistical analysis // Arch Dermatol. 1976. Vol. 112, N 4. Р. 535–542.
- Minić S., Trpinac D., Obradović M. Incontinentia pigmenti diagnostic criteria update // Clin Genet. 2014. Vol. 85, N 6. Р. 536–542. doi: 10.1111/cge.12223
- Haque M.N., Ohtsubo M., Nishina S., et al. Analysis of IKBKG/ NEMO gene in five Japanese cases of incontinentia pigmenti with retinopathy: Fine genomic assay of a rare male case with mosaicism // J Hum Genet. 2021. Vol. 66, N 2. Р. 645–645. doi: 10.1038/s10038-020-00836-3
- Kawai M., Kato T., Tsutsumi M., et al. Molecular analysis of low-level mosaicism of the IKBKG mutation using the X Chromosome Inactivation pattern in Incontinentia Pigmenti // Mol Genet Genomic Med. 2020. Vol. 8, N 12. Р. e1531. doi: 10.1002/mgg3.1531
- Tak P.P., Firestein G.S. NF-κB: A key role in inflammatory diseases // J Clin Invest. 2001. Vol. 107, N 1. Р. 7–11. doi: 10.1172/JCI11830
- Kleinman J.T. Early Wallerian degeneration on magnetic resonance imaging: Underappreciated but highly relevant // Dev Med Child Neurol. 2013. Vol. 55, N 2. Р. 104–105. doi: 10.1111/dmcn.12022
- Salamon S.A., Lichtenbelt K., Cowan F.M., et al. Clinical presentation and spectrum of neuroimaging findings in newborn infants with incontinentia pigmenti // Dev Med Child Neurol. 2016. Vol. 58, N 10. Р. 1076–1084. doi: 10.1111/dmcn.13140
- Lou H., Zhang L., Xiao W., et al. Nearly completely reversible brain abnormalities in a patient with incontinentia pigmenti // Am J Neuroradiol. 2008. Vol. 29, N 3. Р. 431–433. doi: 10.3174/ajnr.A0890
- Kinoshita T., Ogawa T., Yoshida Y., et al, Curvilinear T1 hyperintense lesions representing cortical necrosis after cerebral infarction // Neuroradiology. 2005. Vol. 47, N 7. Р. 647–651. doi: 10.1007/s00234-005-1398-0
- Hauw J.J., Perié G., Bonnette J., Escourolle R. [Neuropathological study of incontinentia pigmenti. Anatomical case report (author’s transl). (In French)] // Acta Neuropathol. 1977. Vol. 38, N 2. Р. 159–162. doi: 10.1007/BF00688564
Дополнительные файлы

